《Organometallic Chemistry》章末总结2
第二章讨论金属有机的基元反应。
2.1 Ligand substitution
配合物配体的交换是非常基础又非常重要的反应,往往是催化循环启动的前置条件。配体交换在机理上可以分为两类,包括 associative ligand substituion 和 dissociative ligand substitution。顾名思义,前者是新的配体结合后原有的配体再离去,后者则是原有配体离去后新配体再结合,分别可以类比 和 机理。
Associative ligand substitution 需要配合物本身具有能够容纳配体的空轨道,因此常见于平面四方形的 型配合物。
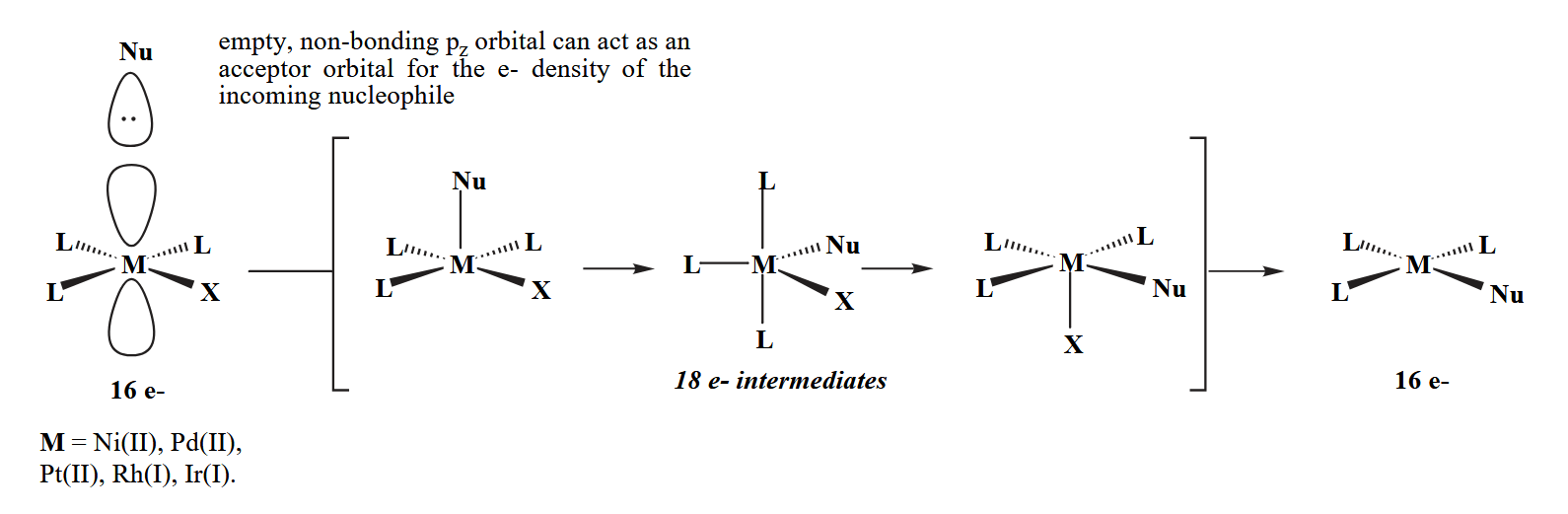
Dissociative ligand substitution 常见于配位饱和的配合物,必须要先解离一个配体才能让另一个配体结合上来。
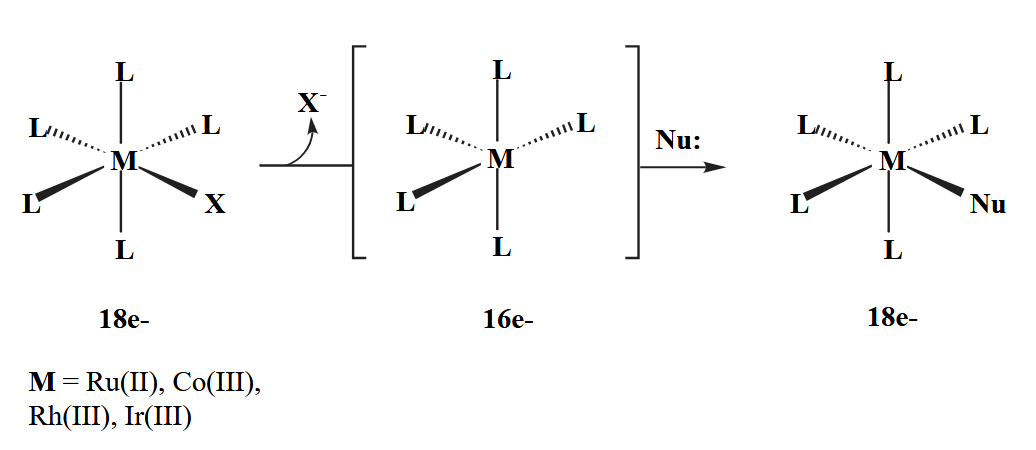
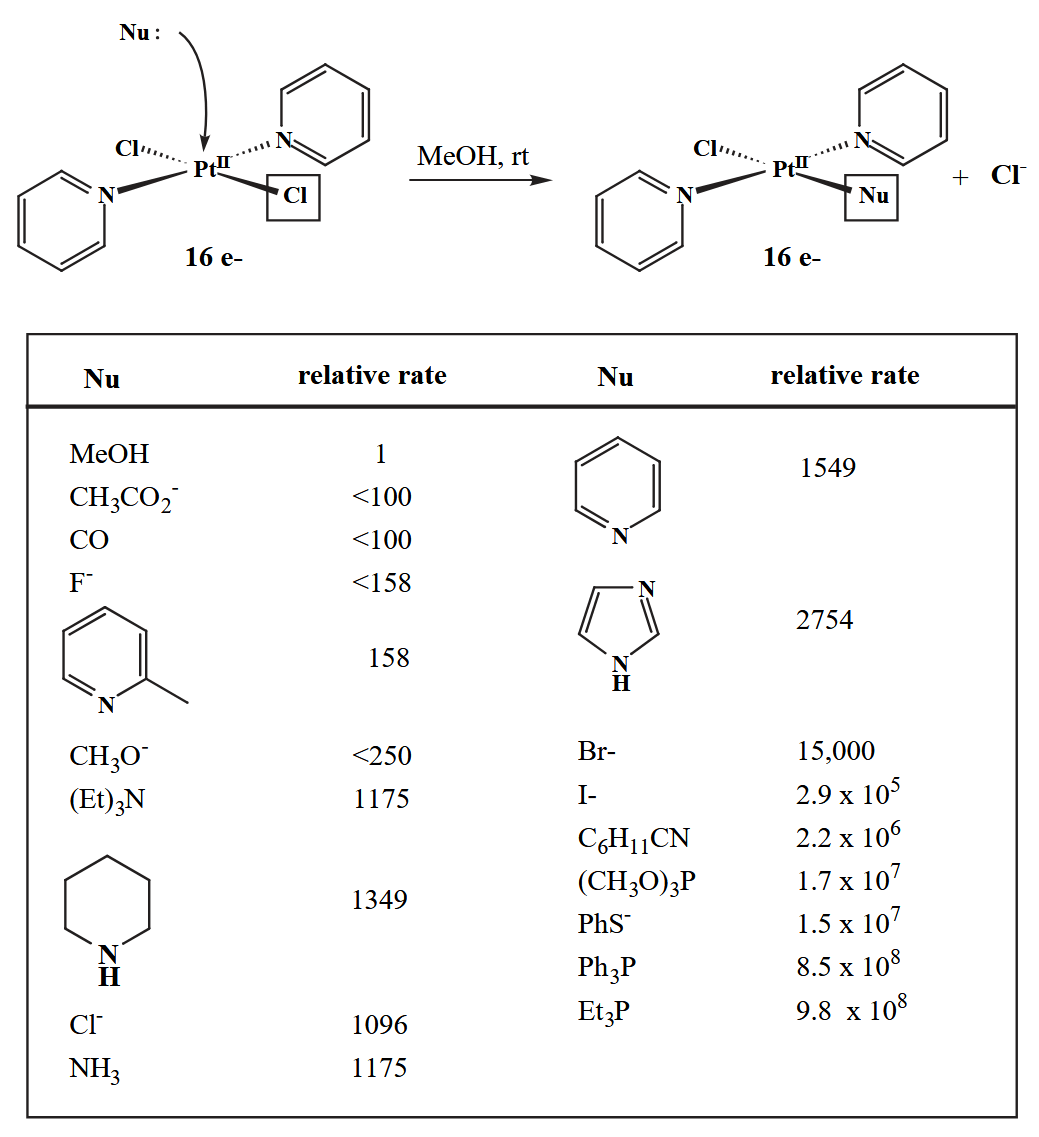
对于 构型的 配合物来说,中心金属本身是软酸,因此倾向于和软碱配体结合,体现在反应速率上就是越软的配体发生 associative ligand substitution 的速率越大。同时位阻也会很大程度上影响配体交换速率。
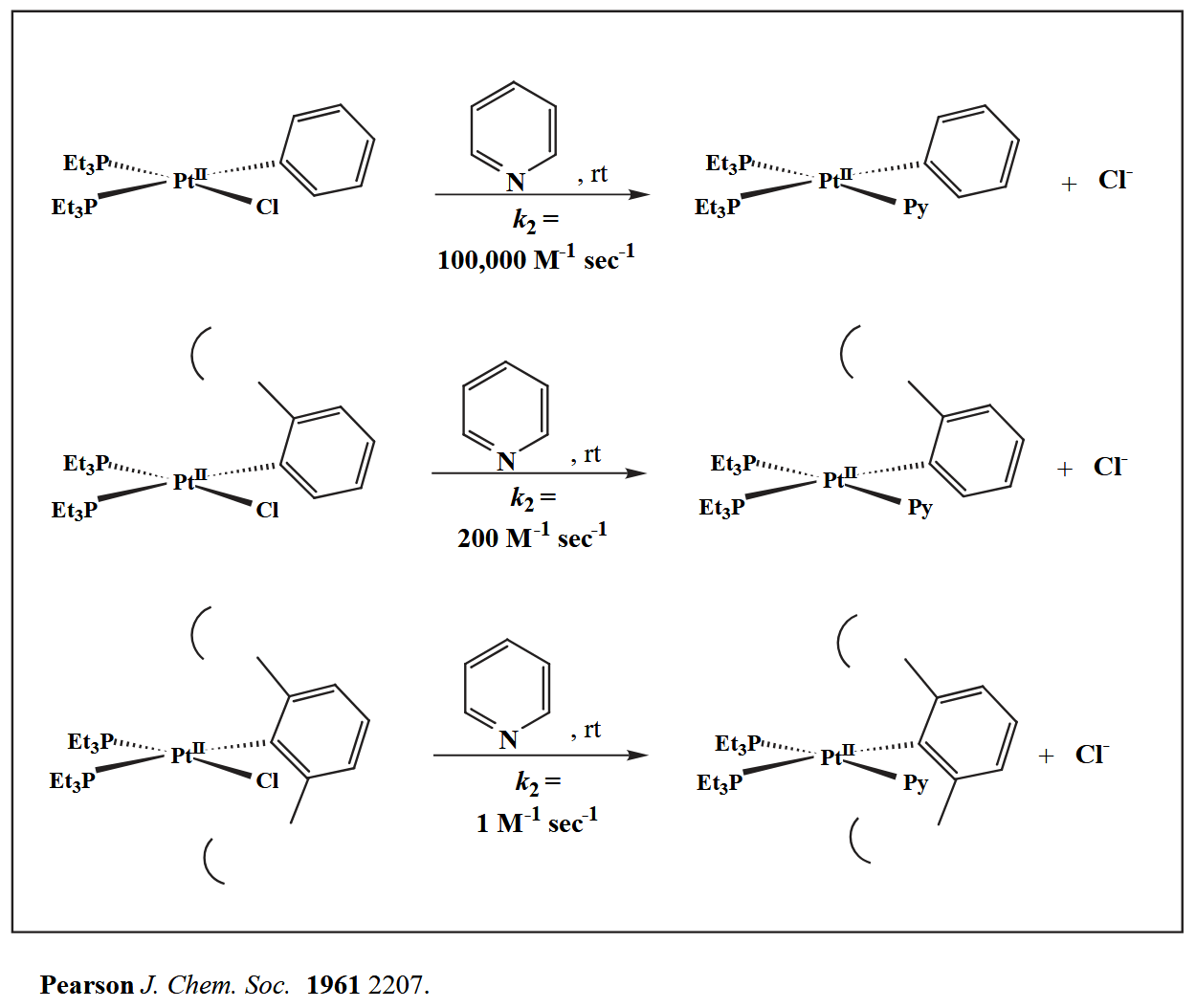
位阻对于配体取代反应的影响有一个很直观的例子可以证明,即下图中的烯烃聚合催化剂。结合大位阻配体的催化剂更不容易发生配体交换的副反应,因此更容易得到高分子量的烯烃聚合物。
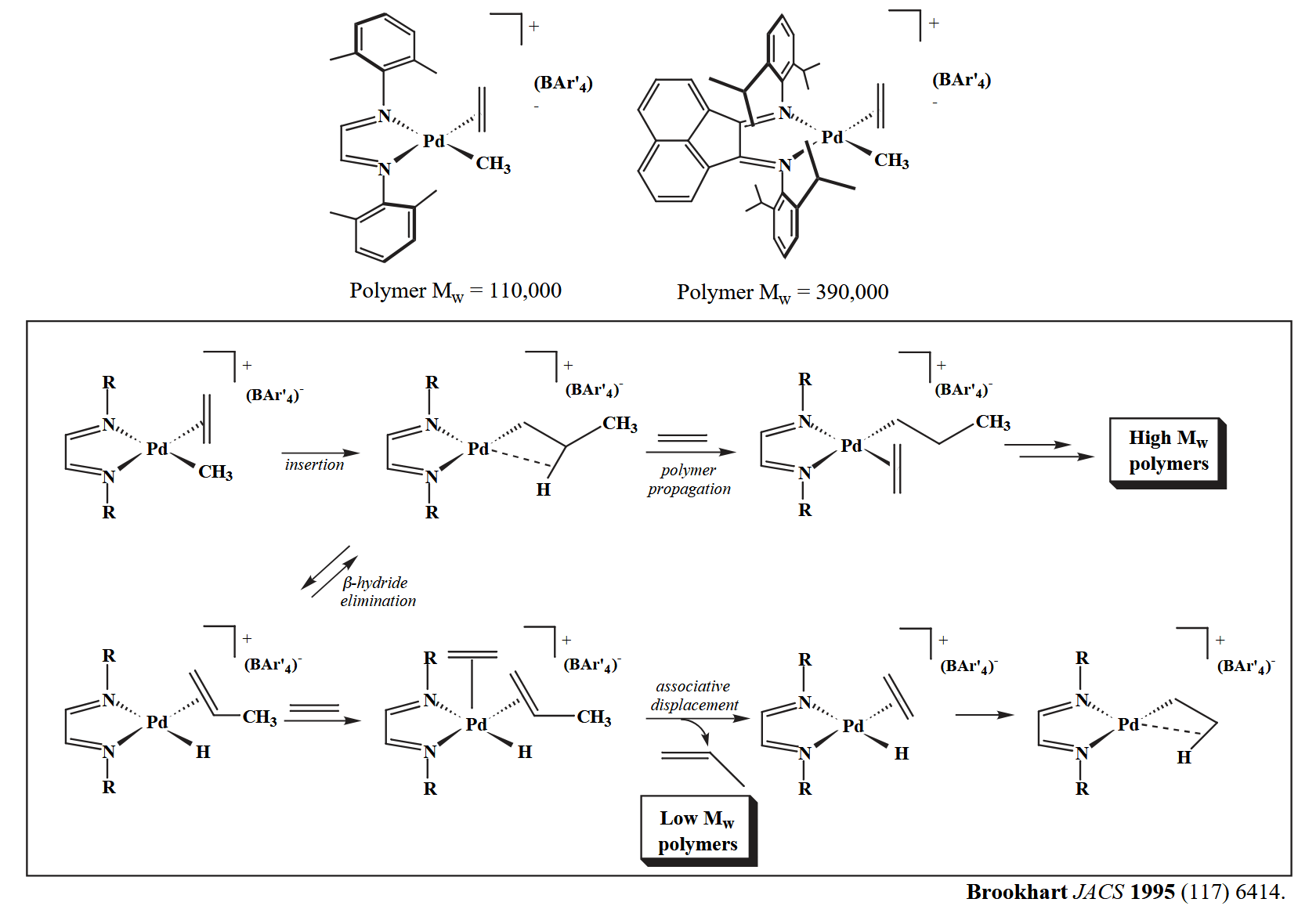
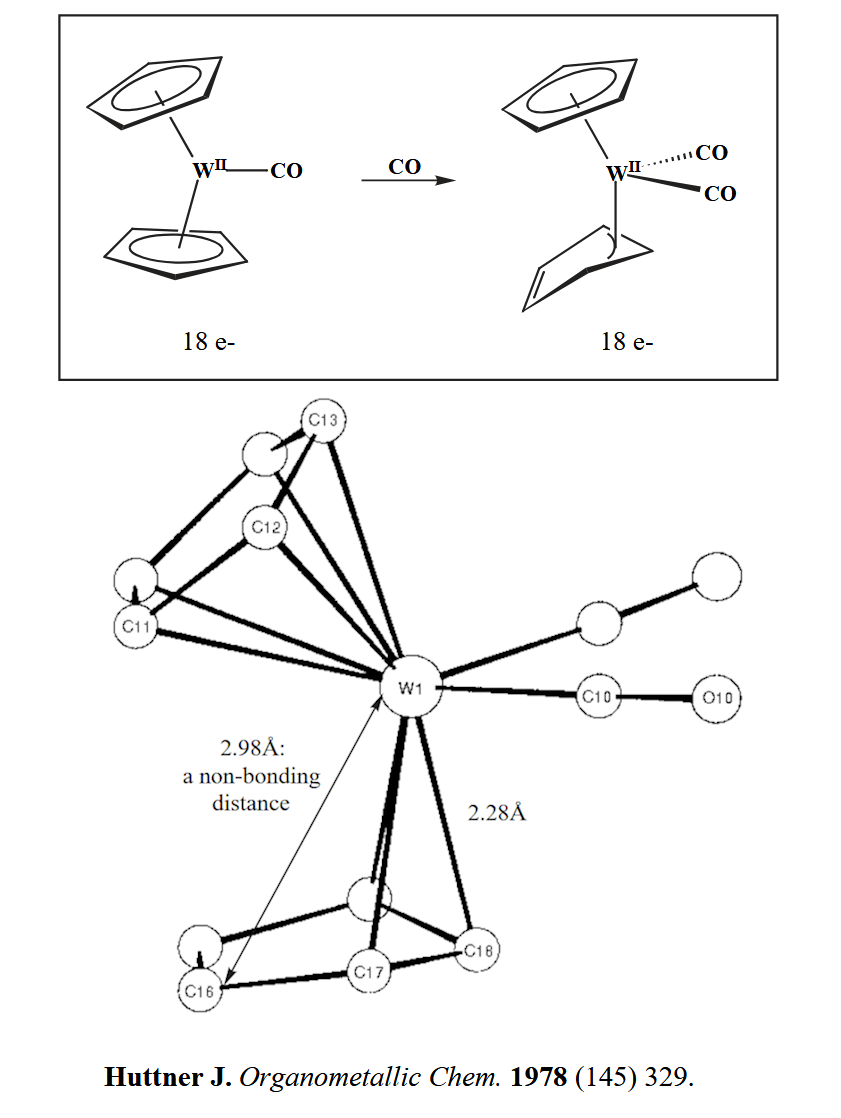
对于芳环配体配合物,有时候这些配合物在电子排列上已经饱和(18e),但是在配体数量上仍可继续结合,因此如果外加配体,可以得到更高配位数的化合物。
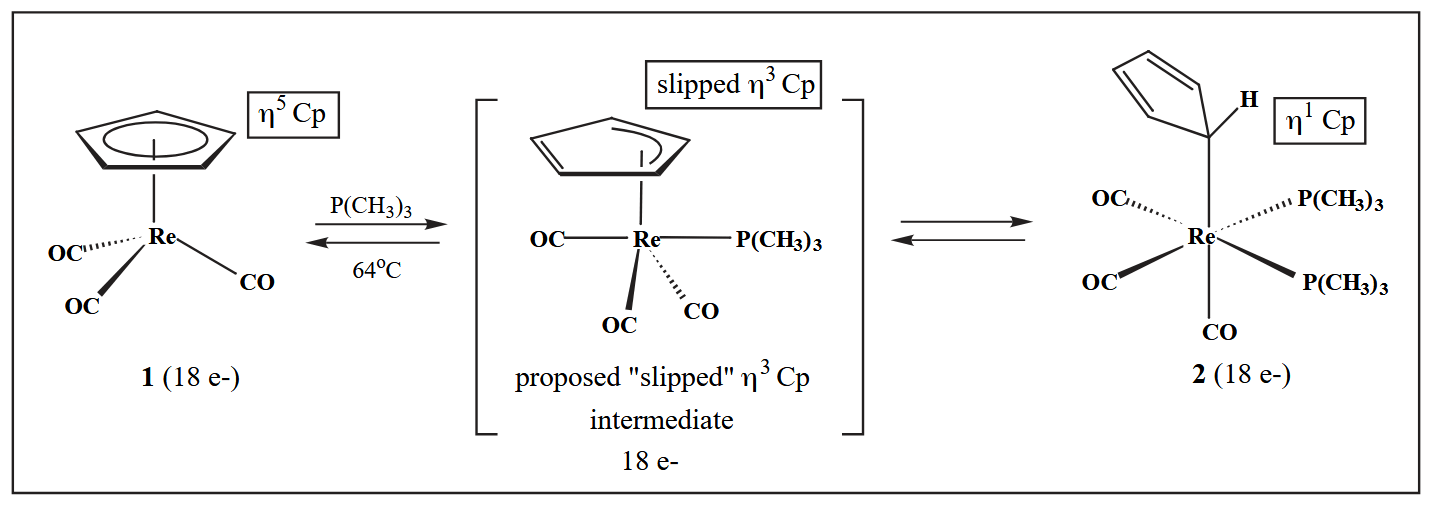
在上图例子中,茂基从 6e 给体最终变成 2e 给体,使中心金属始终保持 18e 构型,Casey 等人推断其经历了一个从 的“滑动”过程。[1]
再看另一个相似的例子:
Dissociative ligand substitution 的第一步是配体的解离,这也是整个反应的决速步(rate-determining step, RDS),影响因素包括电子效应和位阻效应。
对于一个 18e 构型的配合物,离去一个配体相当于破坏了 18e 的稳定构型,因此非常困难。相比之下,从 16e 转化为 14e 就更加容易。
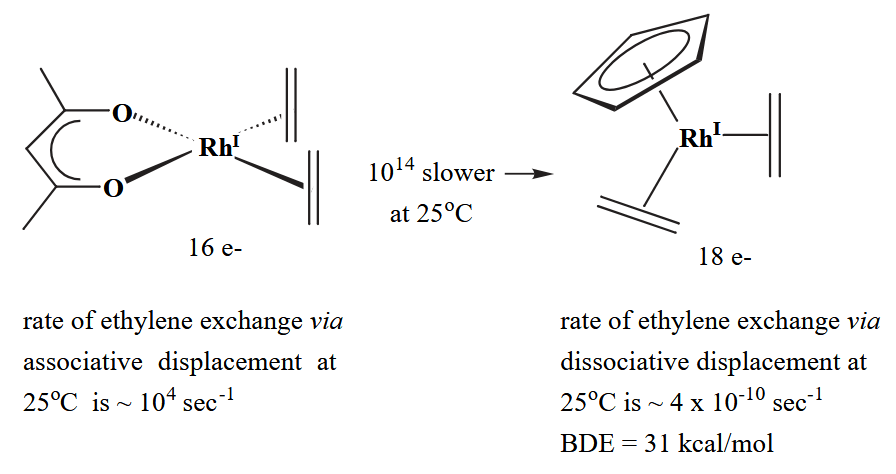
大位阻的配体会削弱配位原子与金属原子的结合,使得配体更易于解离,同时大位阻配体离去后,金属中心也能与反应物接触反应。
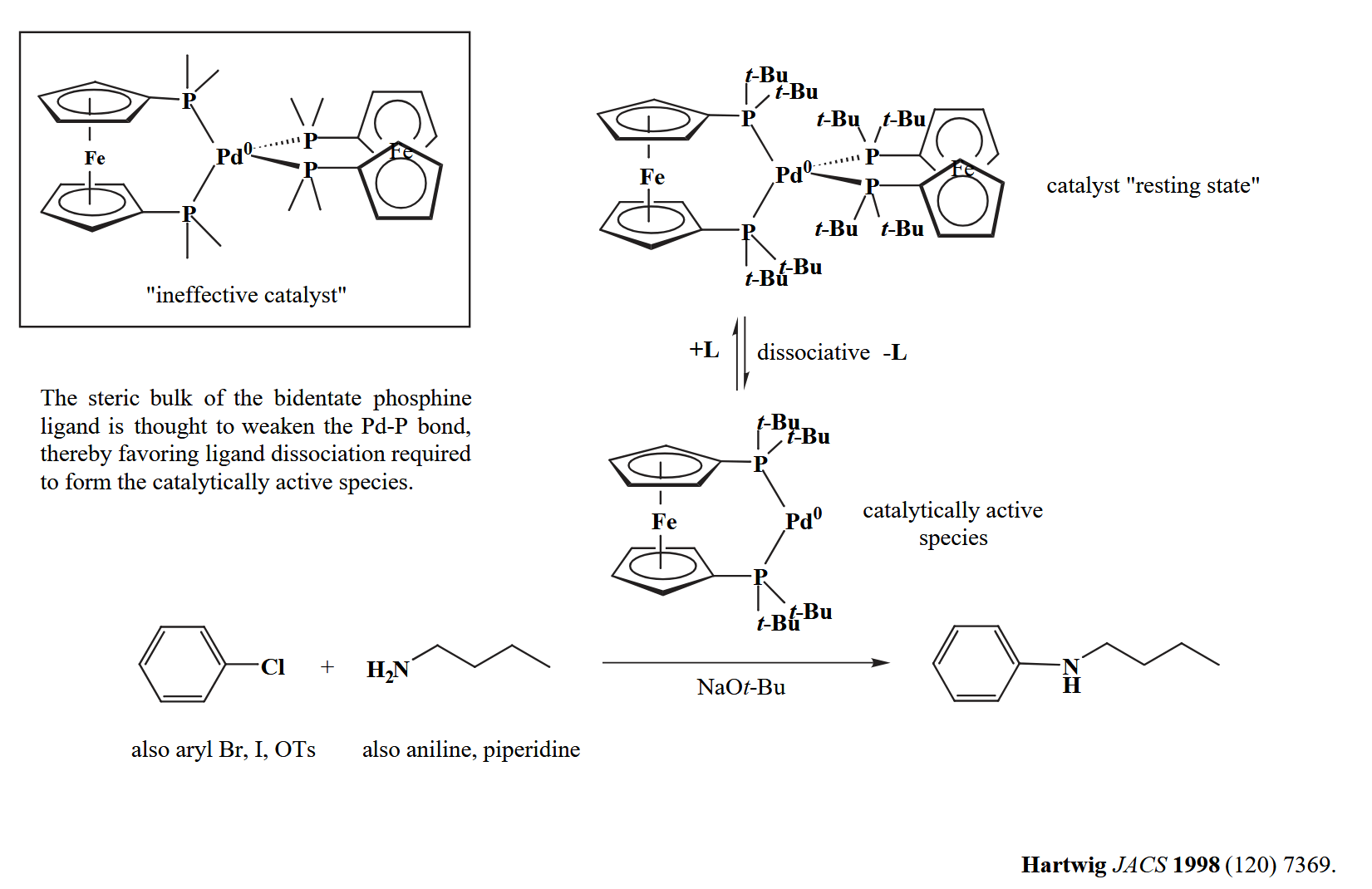
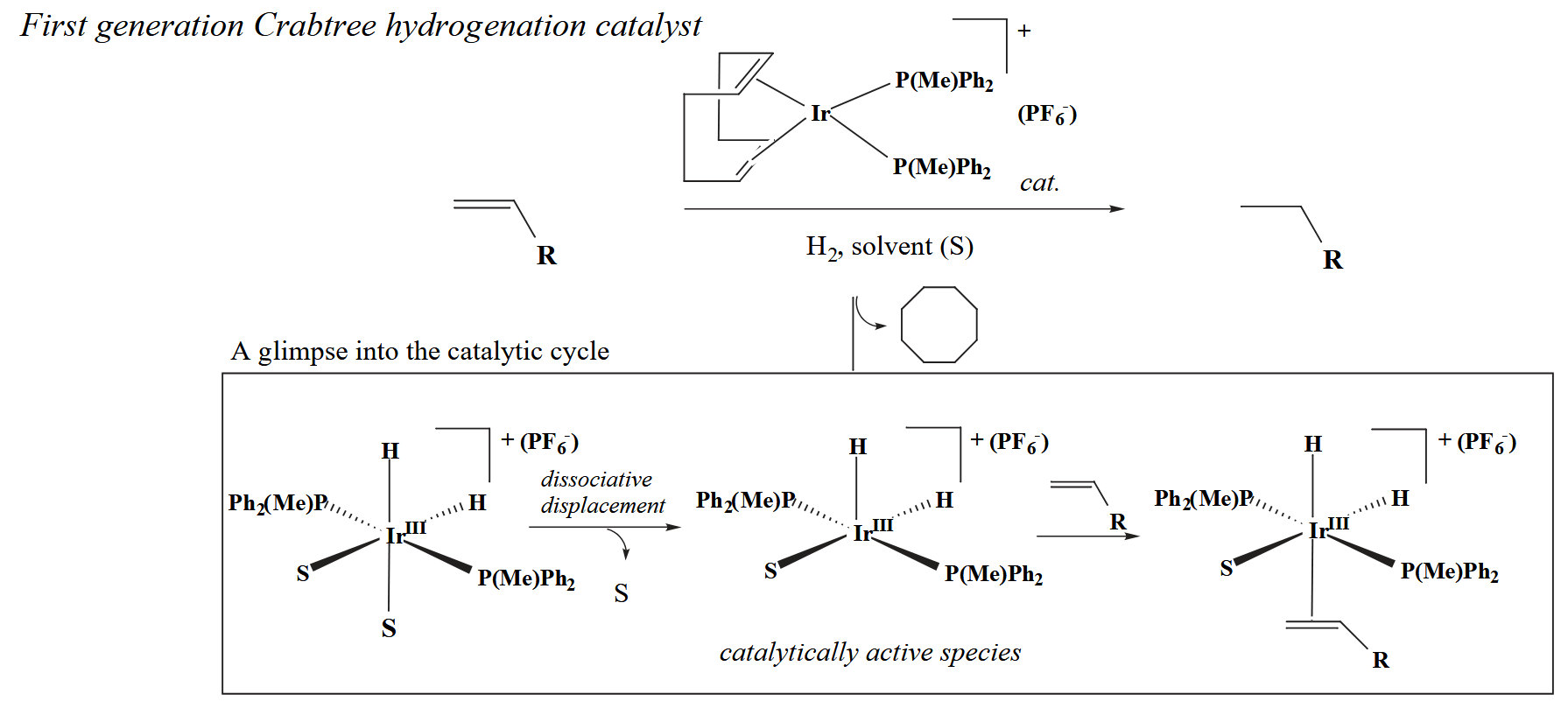
体系中的溶剂往往会与金属配位,因此也会影响到 dissociative ligand substitution 的过程。
2.2 Oxidative addition/Reductive elimination
氧化加成(Oxidative addition, OA)和还原消除(Reductive elimination, RE)是在微观上互为可逆的一对反应,也往往是催化循环的开始和结束。氧化加成会断裂底物的一根 σ-bond 然后形成两个新的 M-L bond,同时金属的氧化态升高2,相应的,还原消除则是断裂两根 M-L bond 接着形成一根新的 σ-bond,同时金属的氧化态降低2。

从上面的描述可以想得到,低氧化态、富电子的金属 + 大极性的亲电底物(高极性的 R-X bond)容易发生氧化加成;高氧化态、缺电子的金属 + 小极性的底物(小极性的 C-C bond)容易发生还原消除,同时还原消除时两个配体需处于顺式构型。
首先来讨论氧化加成,氧化加成的具体机理可能包括三种类别,即协同机理(concerted)、极性机理(nucleophilic)和自由基机理(radical)。一般的规律是协同机理常见于非极性底物;极性机理多见于极性底物;自由基机理则对于极性和非极性底物都有可能出现。
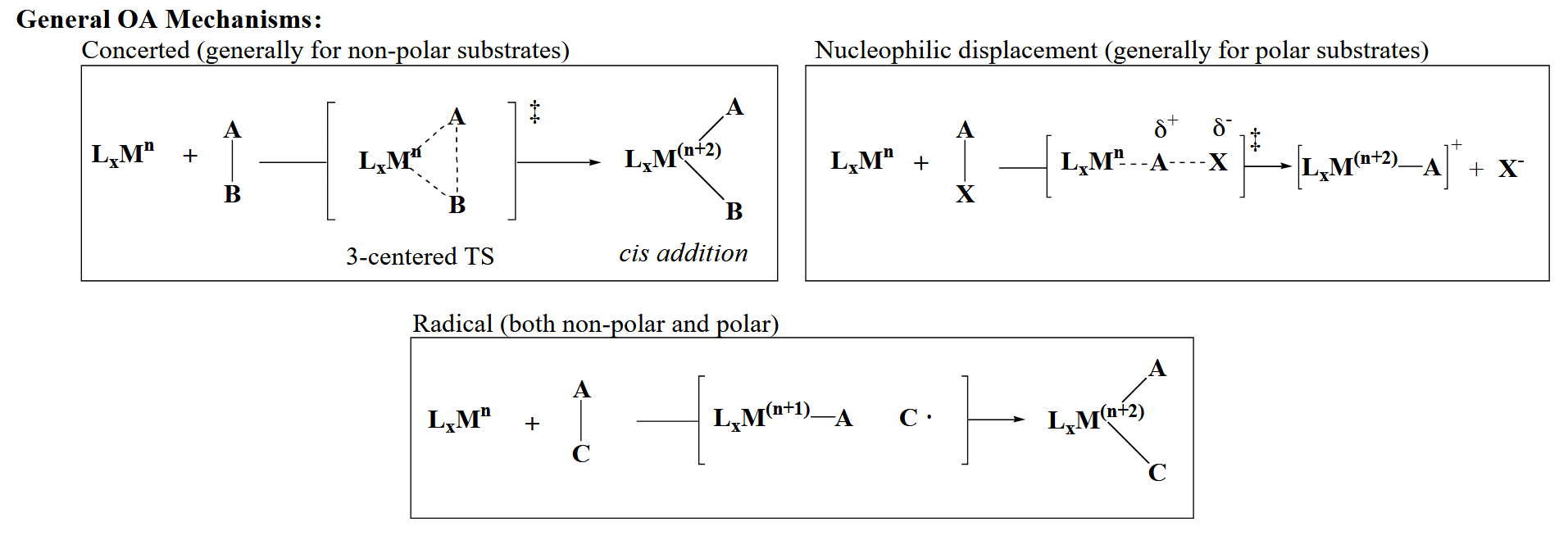
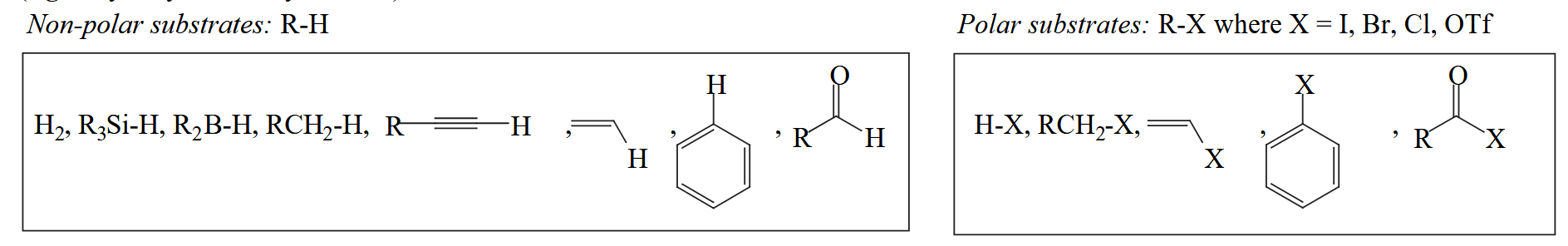
下图中展示的是一些常见底物,通常极性底物更容易发生氧化加成(如芳基卤化物、烯基卤化物等)。
协同机理中典型例子是金属氢化,我们在第一章也提到过氢分子对金属的加成属于 2-way donor-acceptor 类型,与烯烃对金属的配位类似。Kubas 首次分离出了氢分子的金属配合物[2],并发现 H-H 键键长为 0.84Å,比正常 H-H bond 长 20%,这一点很好的证实了金属对氢分子 backbonding 的存在,也是协同机理的一个有力证据。
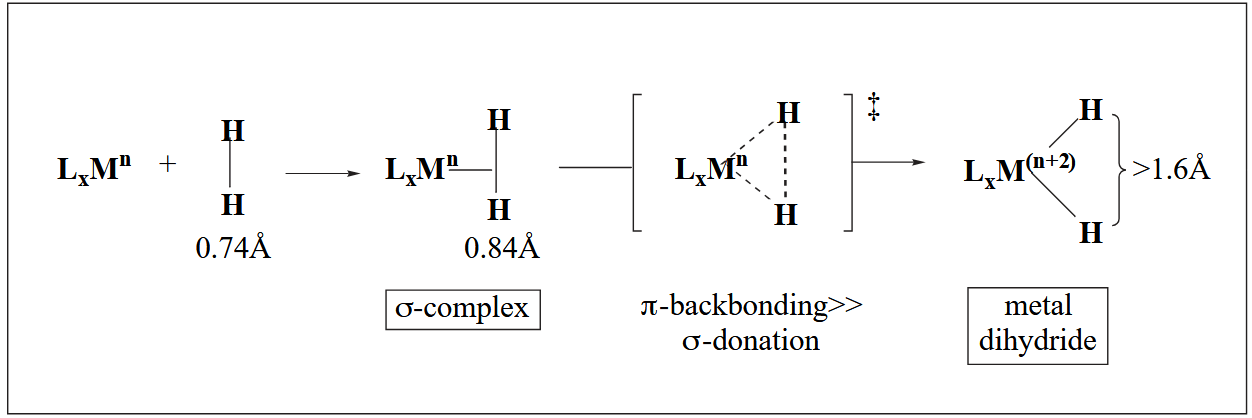
C-H bond 也是一个 non-polar 底物,也会形成类似的 σ-complex。在反应性上,小位阻的 C-H 键和极性大的底物(s 轨道成分更多的杂化碳原子)更易反应,总的顺序是
通常这种非极性底物通过协同机理完成氧化加成,不过 Bergman 等人的机理实验证明,也存在少量的自由基机理产物。
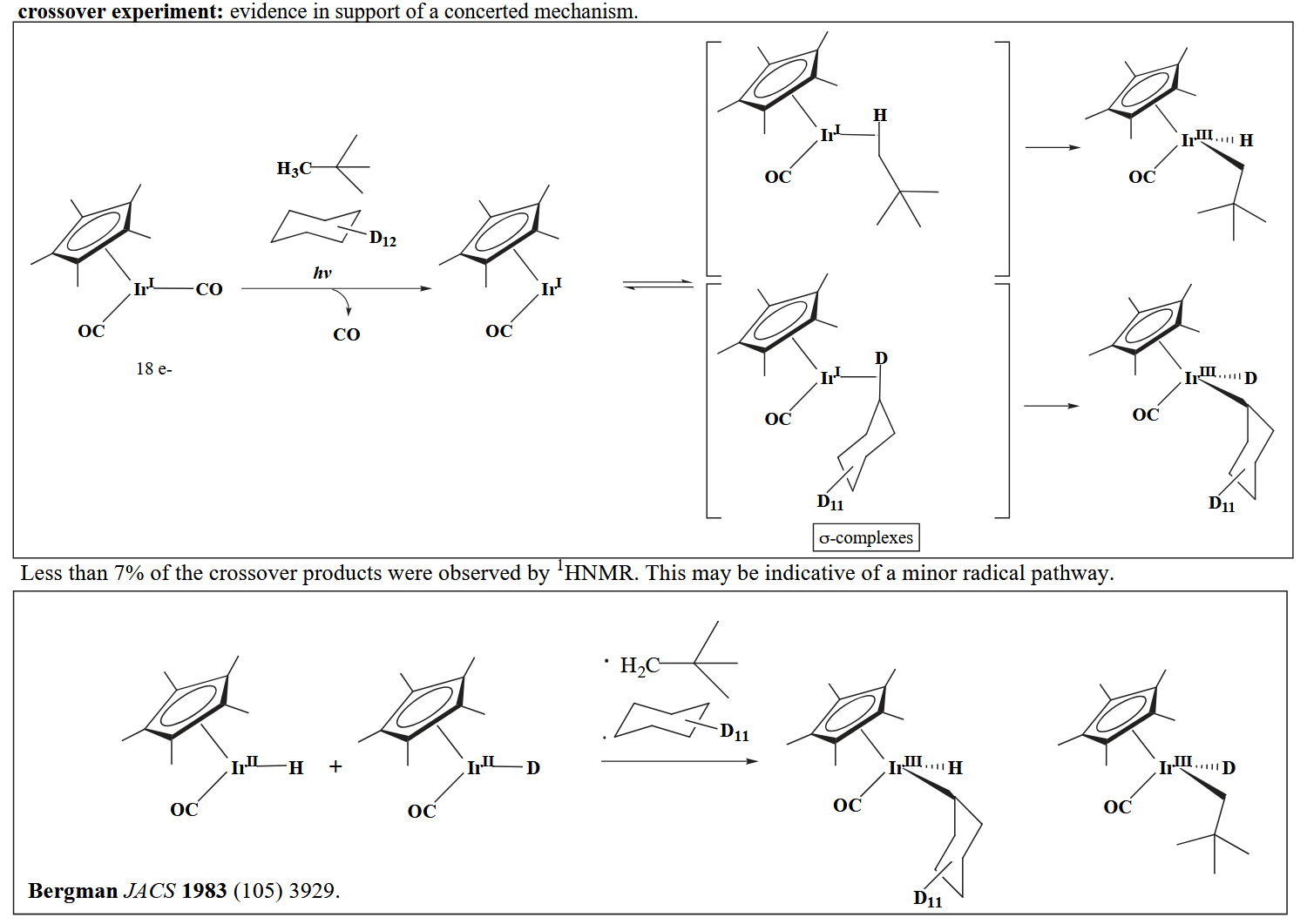
特别注意分子内的 C-H 键氧化加成(C-H 活化)往往存在 agostic interaction,agostic 一词来自于希腊语,有“把某物抓到自己附近”之意,因此国内译为抓氢键,指的就是 M-H-C 之间的弱 3c-2e 作用。
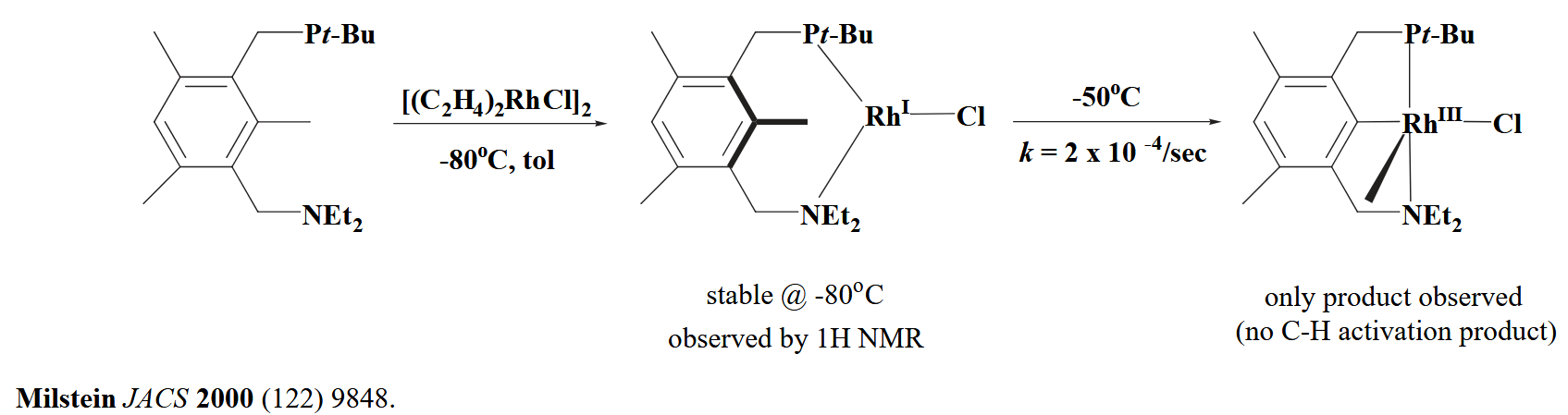
说完了 C-H 键,那么有没有 C-C 键的氧化加成呢?有的兄弟,有的,尽管 C-C 键在位阻上非常不利于发生氧化加成,不过还是有少量的例子存在。例如 Milstein 及其合作者就报道了 Rh 催化下 C-C 键氧化加成的例子。
不过最常见的反应还是对 的氧化加成,烯基卤化物和芳基卤化物都是非常常见的底物。这类型的氧化加成机理仍然存在 concerted process/SNAr-like process/SET process 三种可能,在进一步具体分析之前,我们先引入一个概念:linear free energy relationship (Hammett plots)。
( 简要介绍,更多细节请参考 Carey 的高有 part A。 )
1937年,Louis Plack Hammett 尝试通过不同取代基的芳香化合物的反应来研究反应速率常数和反应平衡常数之间的联系,他在实验中发现速率常数和平衡常数的对数通过数学处理后表现出线性关系,而它们又分别与反应的活化能和 Gibbs 自由能有关,因此称为线性自由能关系(linear free energy relationships, LFER)。
更重要的是,这种线性关系为研究反应过渡态的电性提供了有力工具。对于下式:
式中 为 substituent constant,用于衡量取代基 X 对反应中心施加的极性影响大小。值越正说明基团 X 对反应中心的吸电子效应越强,越负则说明基团 X 对反应中心的给电子效应越强。
式中 为基团 X 取代时反应的平衡常数与无基团 X 取代(或者说 X=H 时)的平衡常数比。而 作为二者的比例系数,其正负就反映出取代基电子效应对反应的影响。总结来说: 为正值说明反应会被吸电子基团促进,因此反应过渡态是带负电; 为负值说明反应会被吸电子基团抑制,因此反应过渡态是带正电。下面是两个例子:
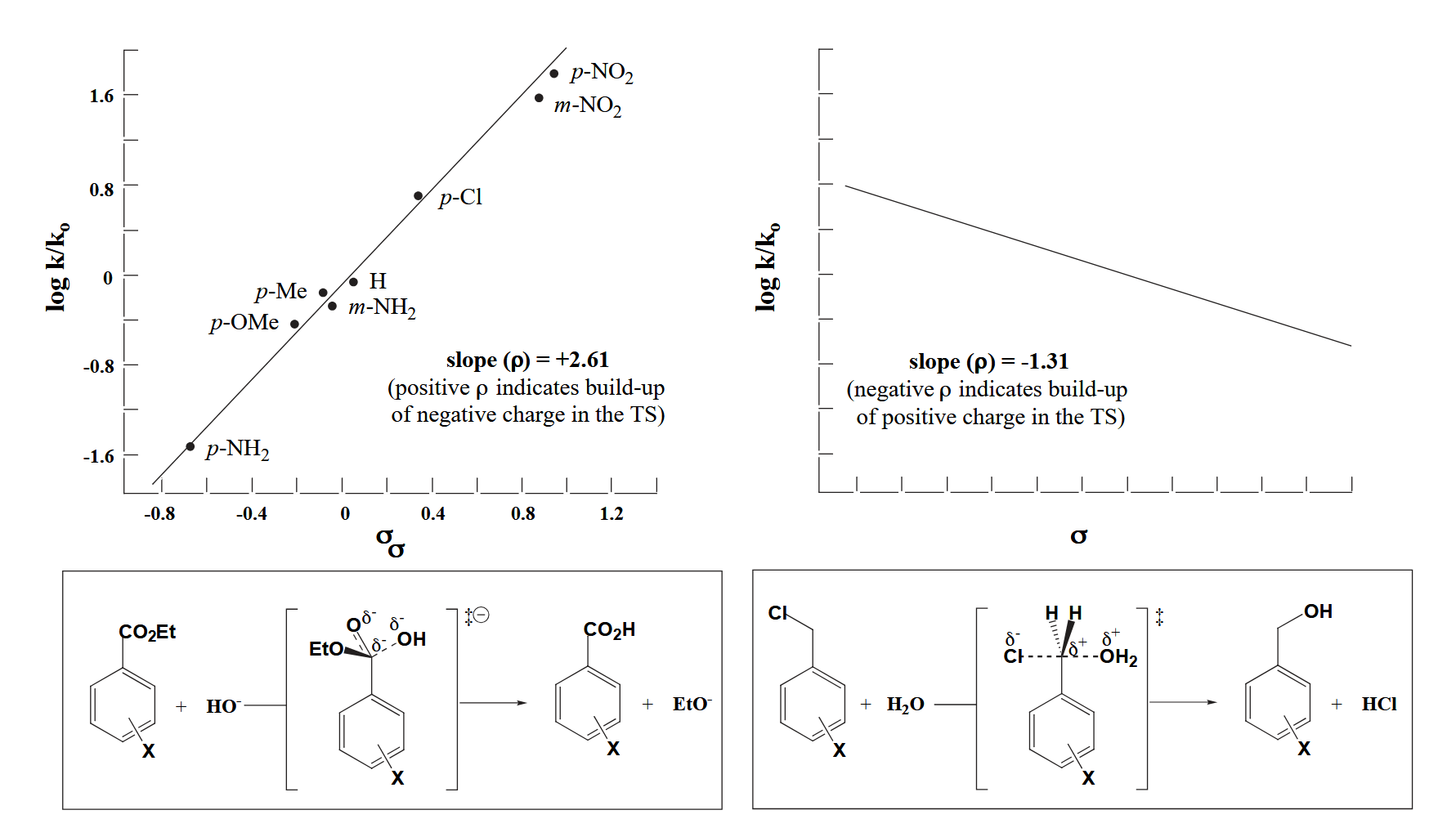
那么言归正传,我们用 LFER 关系分析一下氧化加成反应。
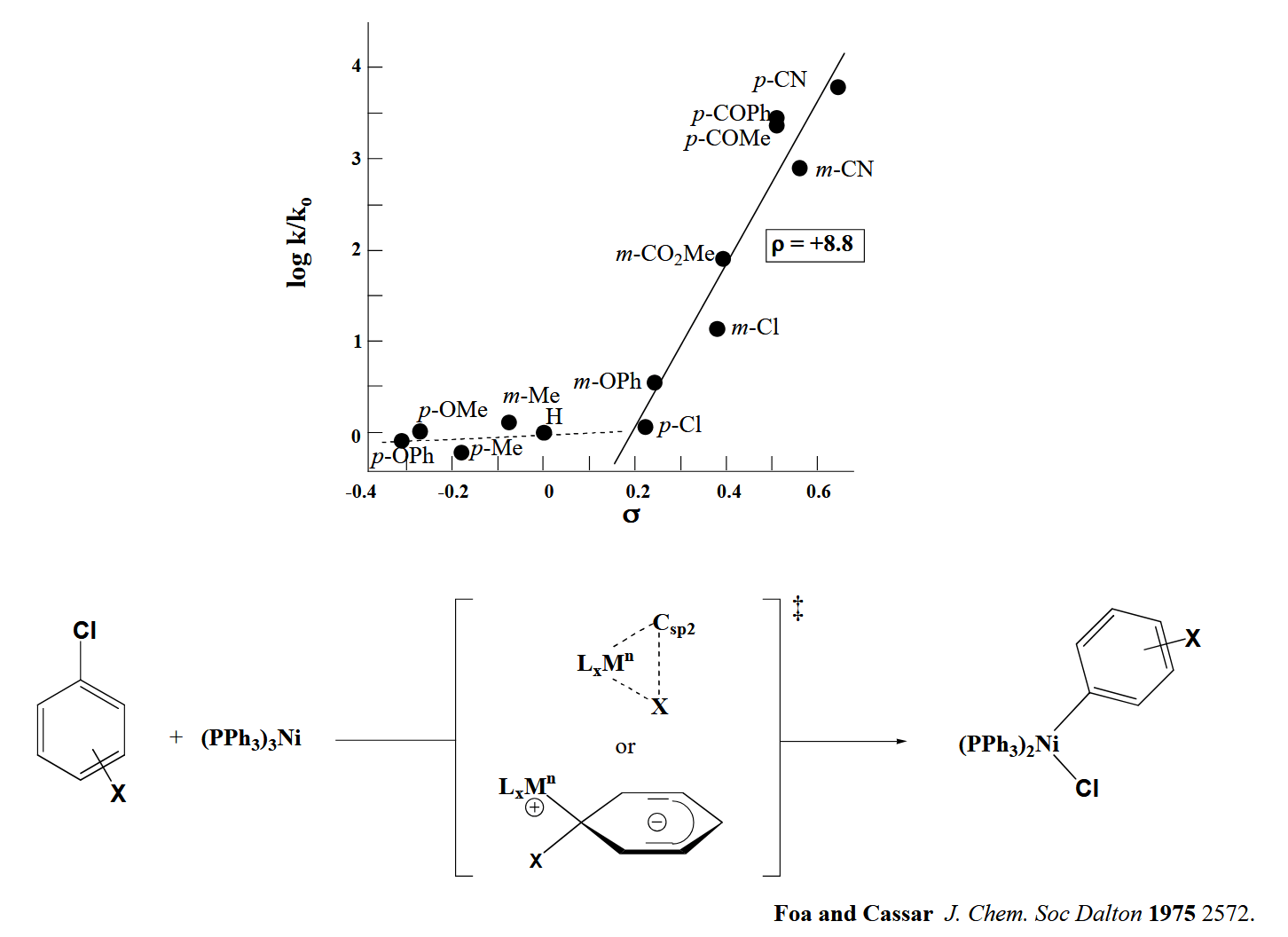
发现当取代基为吸电子基团时, 明显为正值;当取代基为给电子基团时, 为正值但值很小,作者推测这类反应可能经历一个非对称的三中心协同过渡态,而当吸电子基团存在时则经历一个类似芳香亲核取代过程的过渡态。
最后讨论的 bond 氧化加成中有一个特别的反应需要注意,也就是烯丙基卤化物的氧化加成反应,对于这种极性底物的氧化加成通常都是类似 的机理,其中会包括构型的翻转,而金属催化的 反应则会发生两次构型翻转(double inversion),最终得到构型保持的产物。
看下面这个例子:
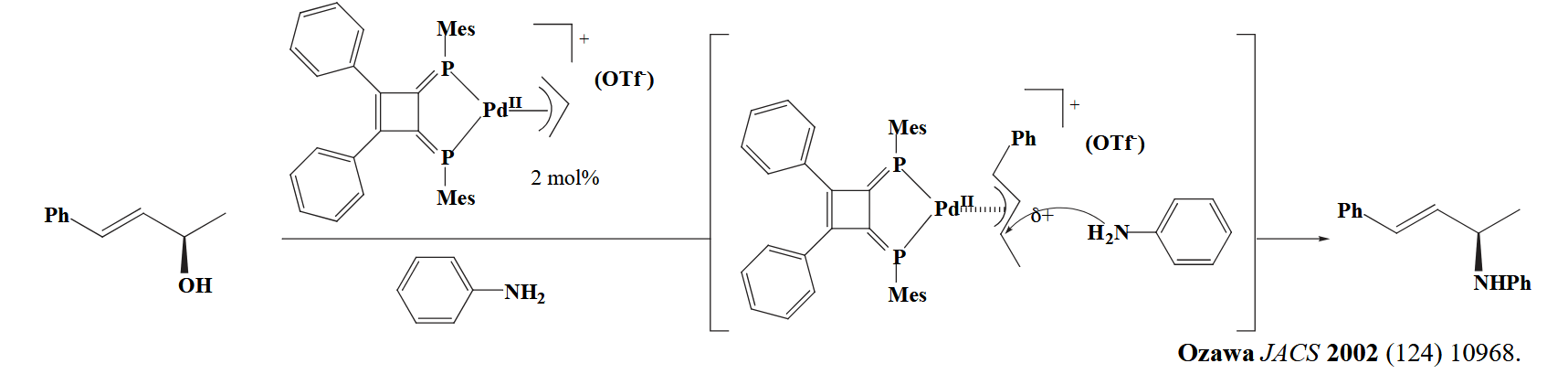
然后是还原消除,作为催化循环的最后一个反应,还原消除的发生意味着偶联产物的生成。还原消除要求两个目标基团处于顺式(-cis)构型,具有更好轨道重叠的基团更容易发生还原消除。
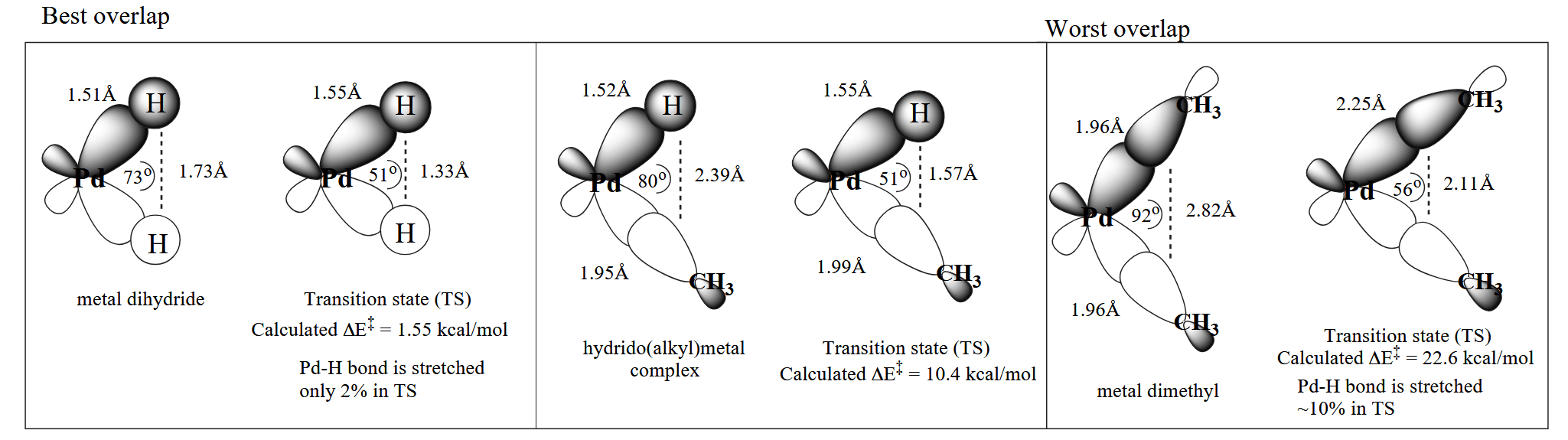
促进还原消除的因素可以从三个方面考虑:增大配体的 bite angle、增加金属中心的亲电性以及增进配体解离。
配体的 bite angle 可以从下图直观理解,可以理解为双齿配体配位原子 X 与金属 M 形成的 。易于理解,bite angle 越大则对位的两个准备发生还原消除的基团在空间上相隔的就越近。
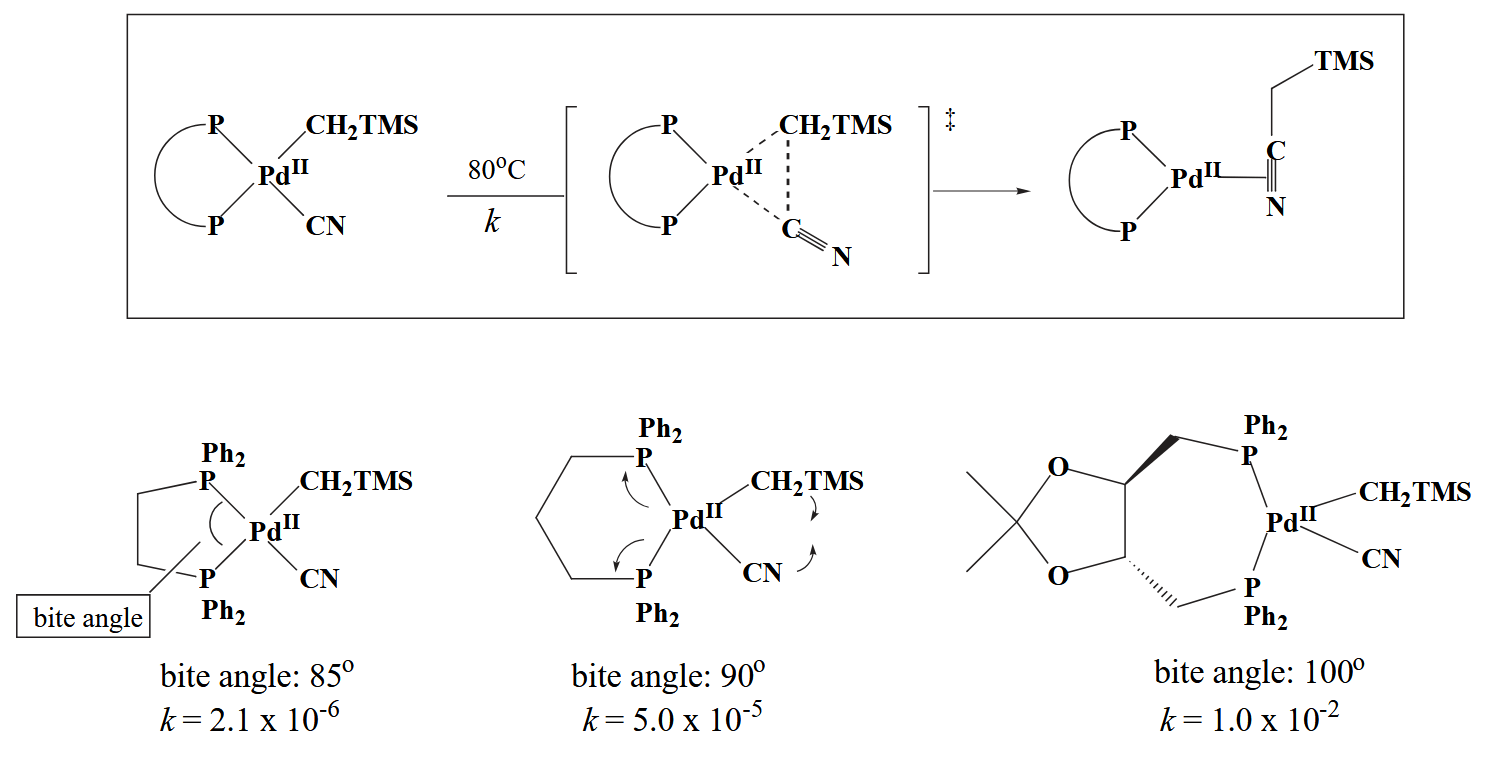
增强金属中心的亲电性可以选择合适的金属以及配体,另外就是利用 π-acid effect,下面是一个例子。
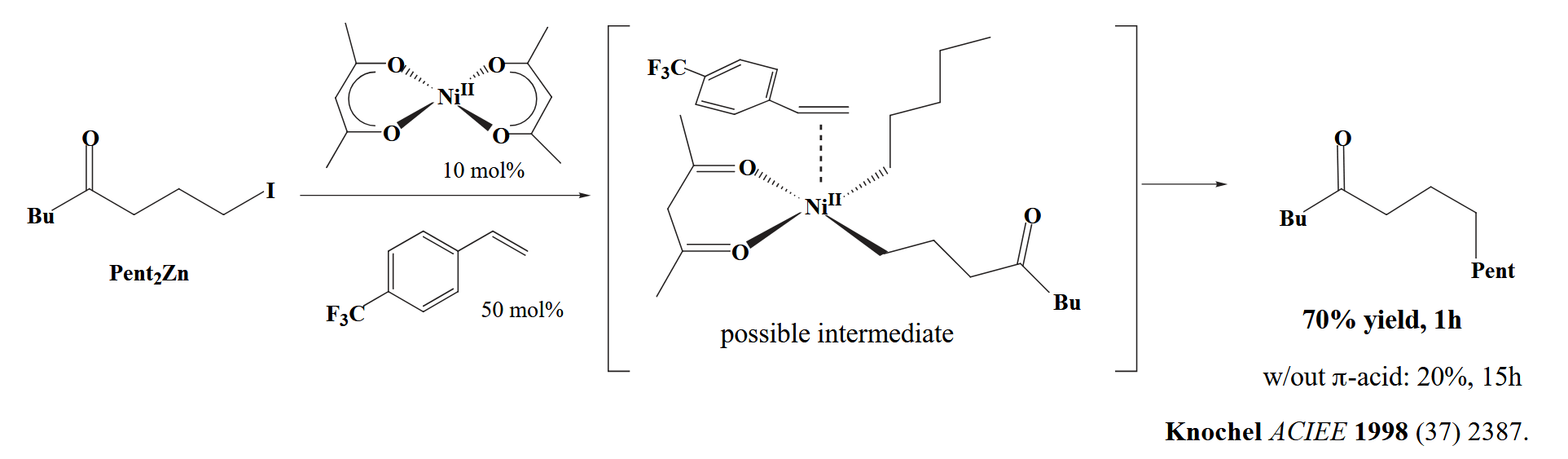
最后,可以让配体更容易解离,例如 C-H 产物比 C-C 产物更容易通过还原消除生成,因为 H 原子的球形 s 轨道的重叠更好。
2.3 Transmetalation
转金属,顾名思义就是两个金属所连基团发生了转移,通过一个类似复分解的过程得到新的金属复合物,在此过程中不会发生金属氧化态的变化。
转金属过程是一个可逆过程,其平衡倾向于生成更稳定的 M-X bond,由于金属通常是小电负性的,所以平衡倾向于生成 more ionic M-X bond,转金属通常是催化循环中间的一步。
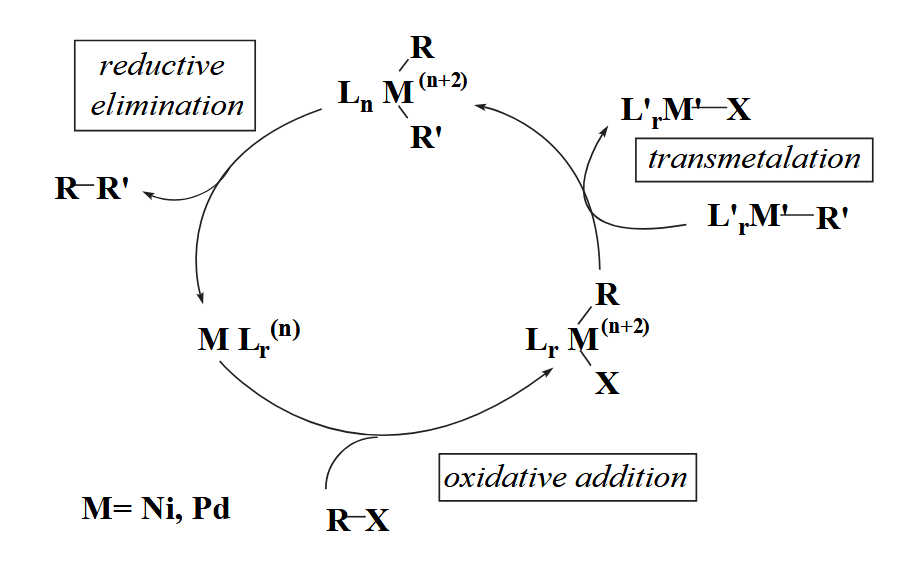
许多过渡金属催化的偶联反应的催化循环往往大同小异,其核心差异就在于转金属一步中金属/配体/底物上的个别不同,见下图(图中比较典型的人名反应会在下一章分别介绍)。整体上来看,不同 R 基团的转金属速率表现为炔基 > 芳基,烯基 > 烷基。
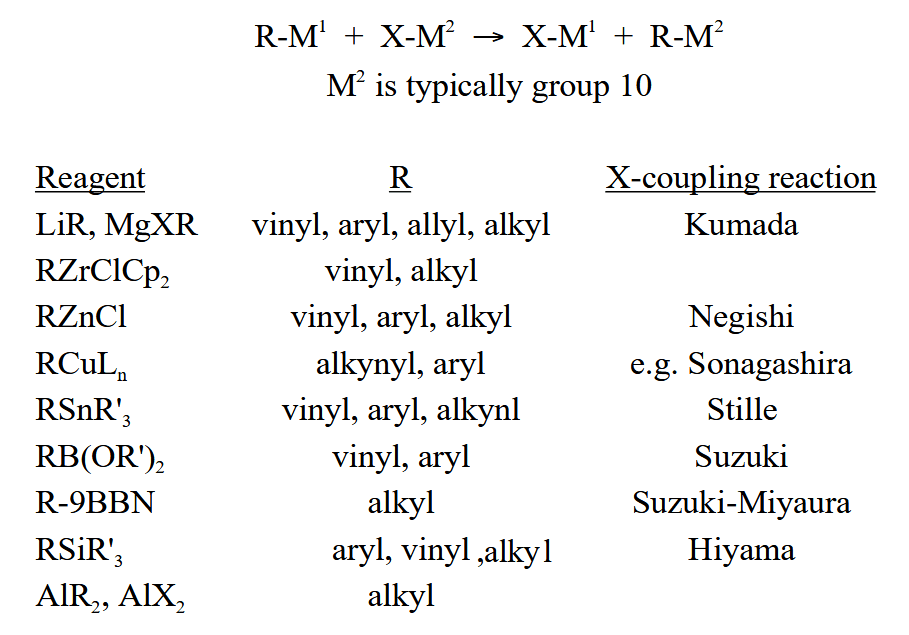
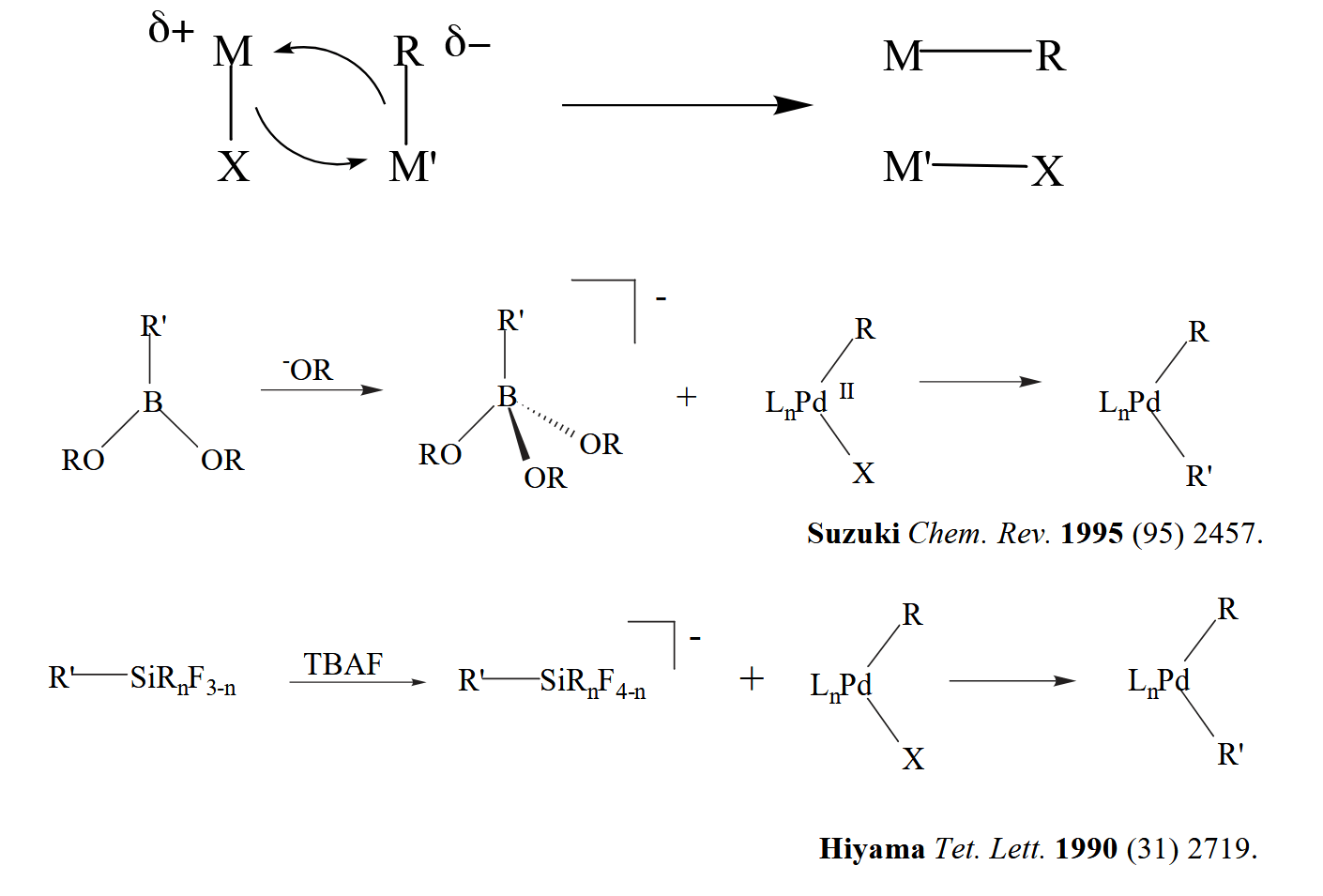
对转金属过程的研究相对较少,反应物极性的差异可能会导致不同的机理,例如协同机理或者极性机理。增强转移集团 R 的负电性或增加催化金属的正电性有利于促进转金属发生。典型例子如 Suzuki 偶联中需要加入碱,以及 Hiyama 偶联中加入 能促进反应。
2.4 Migratory insertion/ β-Elimination
Migratory insertion 和 β-Elimination 是一对互为可逆的反应,表现为金属和连接基团对含 π 电子的物种插入/消除,通常是催化循环中间的一步,并且与反应产物的形成息息相关。
一个很好的例子是 Ziegler-Natta 催化剂催化烯烃聚合的反应,其主要过程就是反复地迁移插入,从而不断延申碳链形成大分子量的聚合物。Cossee-Arlman 最早提出了一种可能的聚合机理,其通过一个四元环协同过渡态。
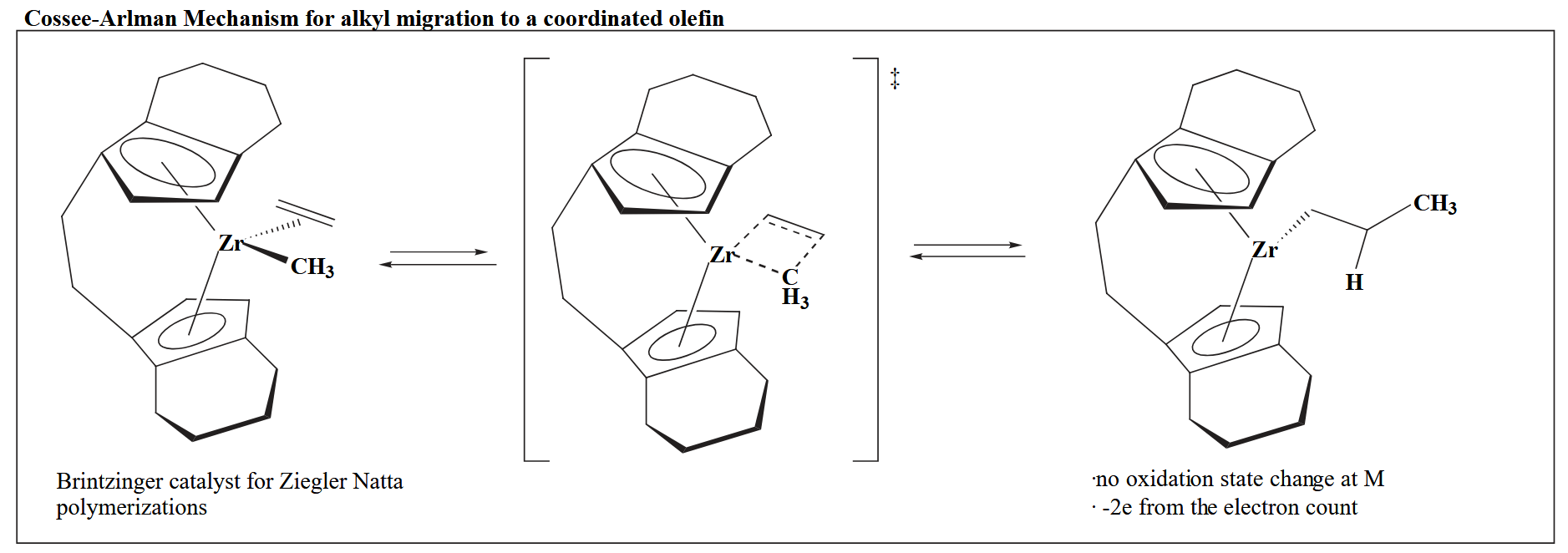
反应是可逆的,因此存在一个平衡,对于金属烷基化合物,反应倾向于生成插入产物,而对于金属氢化物,反应平衡则倾向于消除产物。因此也说明迁移插入反应很容易在平衡控制下生成热力学稳定的最终产物。
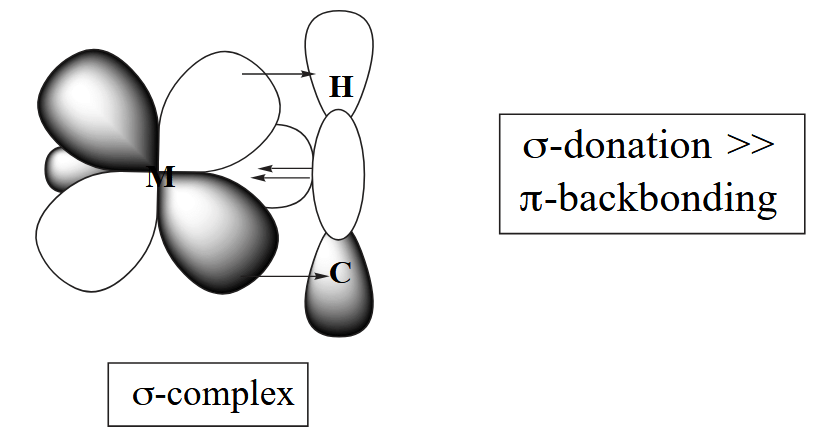
由于反应经历相同的过渡态,因此想要发生 β-elimination 需要让 M-C-C-H 四个原子处于共面,才能形成四元环过渡态,同时在形成 σ-complex 中间体时应该是 σ-donation >> π-backbonding,即存在明显的 agostic interaction。
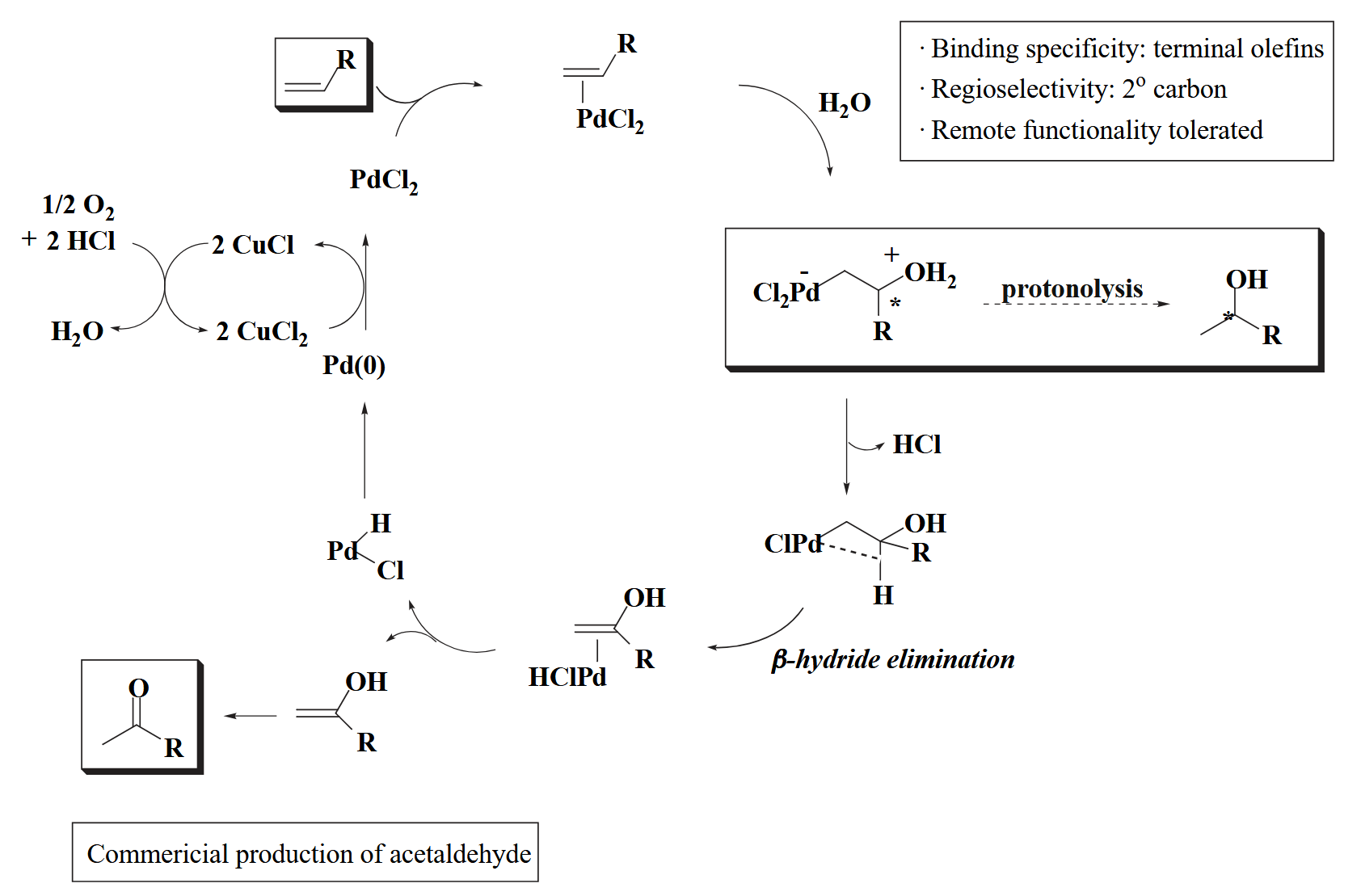
一个例子是 Wacker 氧化,用于商业化的制造乙醛。
对于迁移插入反应,需要特别注意的一个例子是 CO 的迁移插入,与金属相连的基团迁移到羰基碳上,形成羰基化合物。
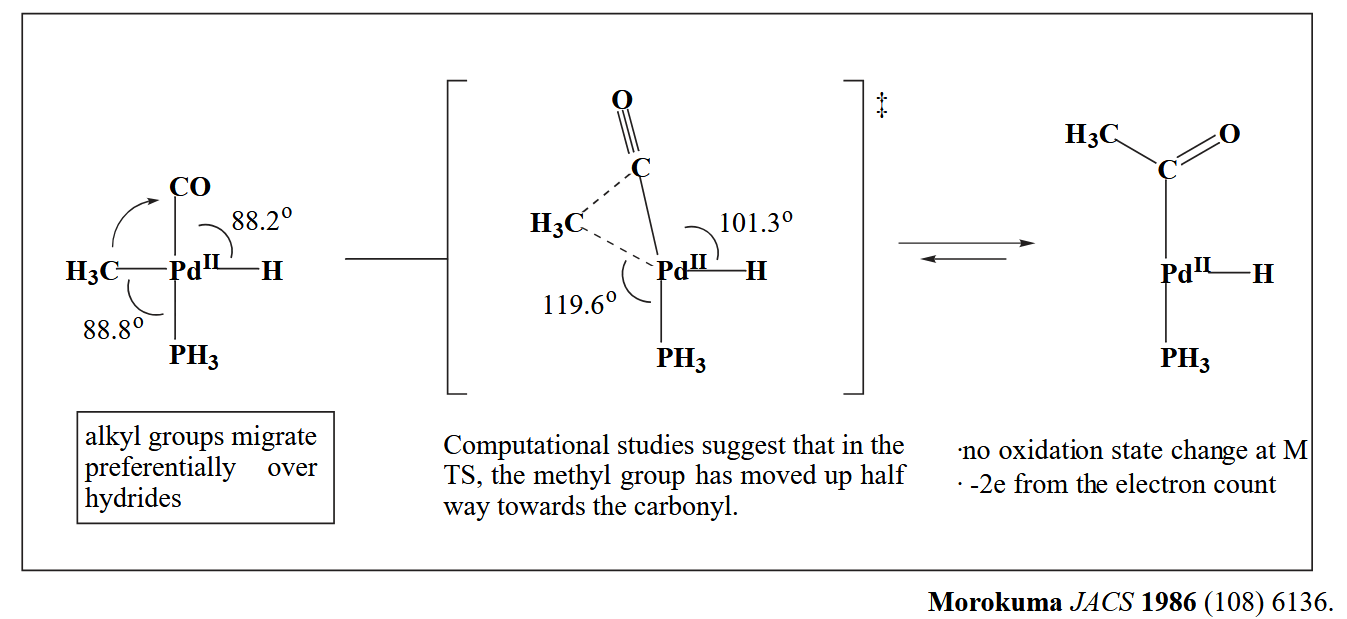
Yamamoto 等人的机理实验证明是烷基发生迁移,而非 CO 的迁移。
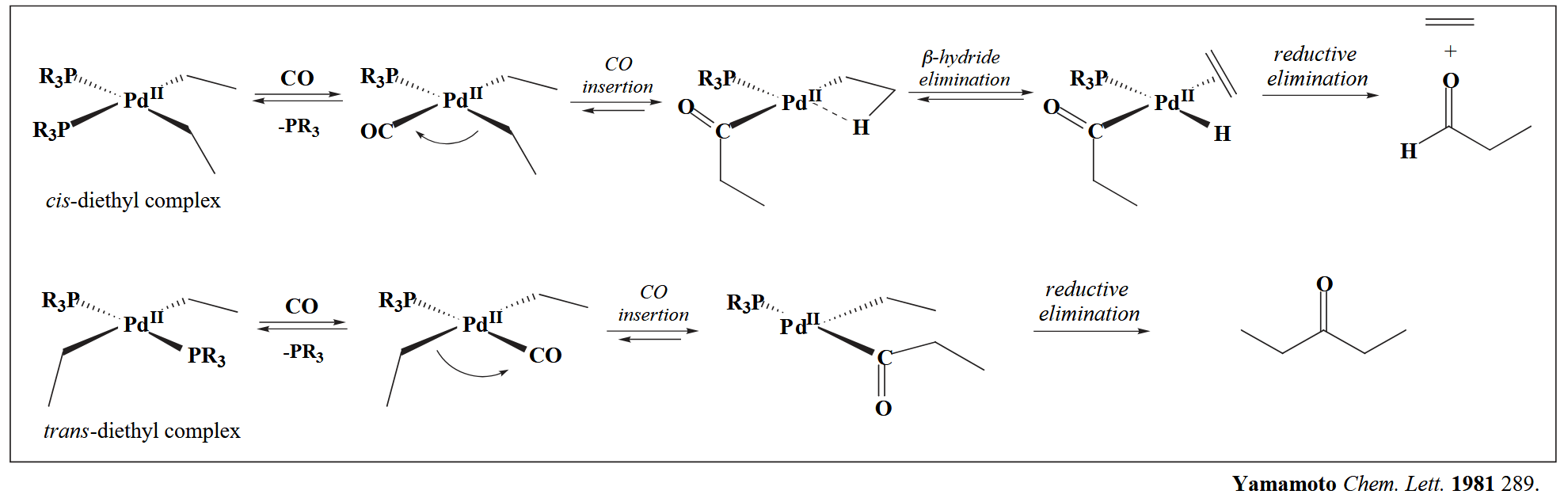
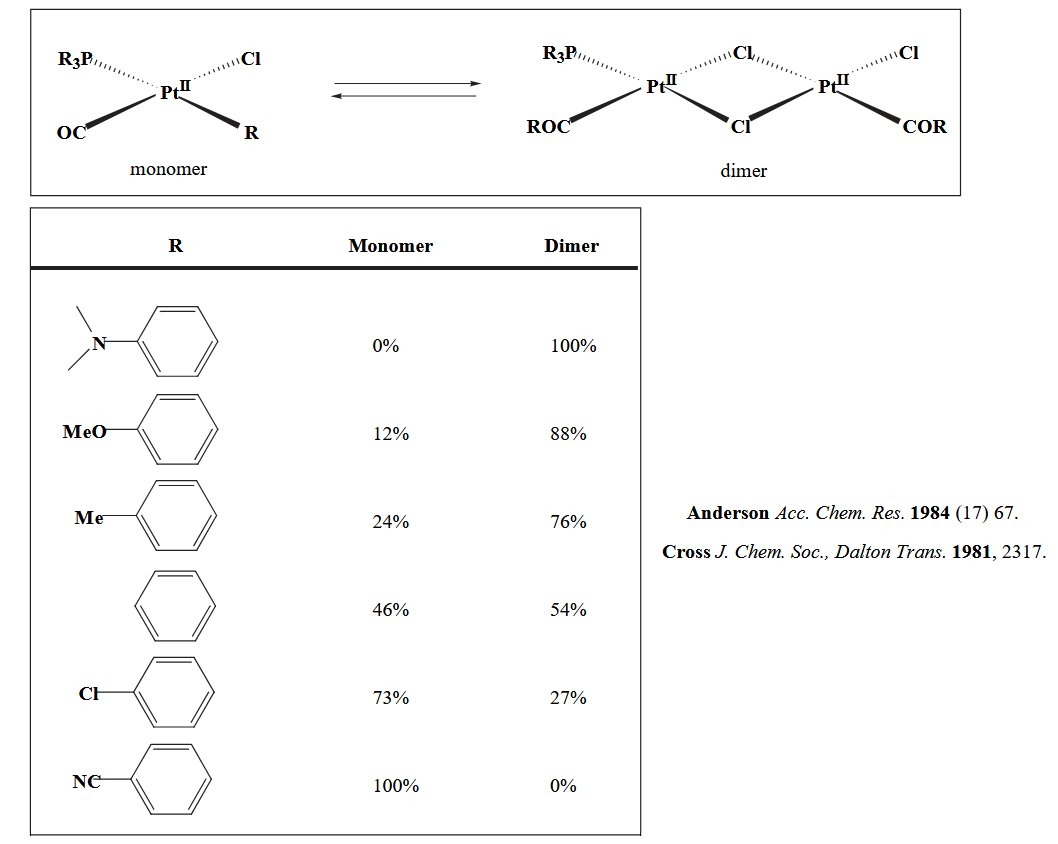
Anderson 等人以及 Cross 等人的机理实验证明给电子能力强的基团更容易迁移。
2.5 Summary
本节主要讨论了常见的几种基元反应,大多数金属有机催化反应都是由这几种最基础的基元反应组合而成,掌握了这几个基本反应模式就能够看懂绝大多数的反应机理。下一节我们将会讨论几个最典型的人名反应。
2.6 reference
- 1.Casey, C. P.; O'Connor, J. M.; Jones, W. D.; Haller, K. J. Intermediates in the Associative Phosphine Substitution Reaction of (.Eta.5-C5H5)Re(CO)3. Organometallics 1983, 2 (4), 535–538. ↩
- 2.Kubas, G. J. Molecular Hydrogen Complexes: Coordination of a .Sigma. Bond to Transition Metals. Acc. Chem. Res. 1988, 21 (3), 120–128. ↩