《Organometallic-Chemistry》章末总结3
本章着重讨论过渡金属催化(以 Pd 为例)的偶联反应中最常见的几个反应。
3.1 Overview
我们下面将要讨论的几个反应的总体反应模式大同小异,主要区别是使用的主族金属不同,不同的金属导致了不同的反应性,但是机理不会有大的改变。
反应的一般机理见下图,主要包括 1) 催化物种的生成、2) 氧化加成、3) 转金属、4) 还原消除。
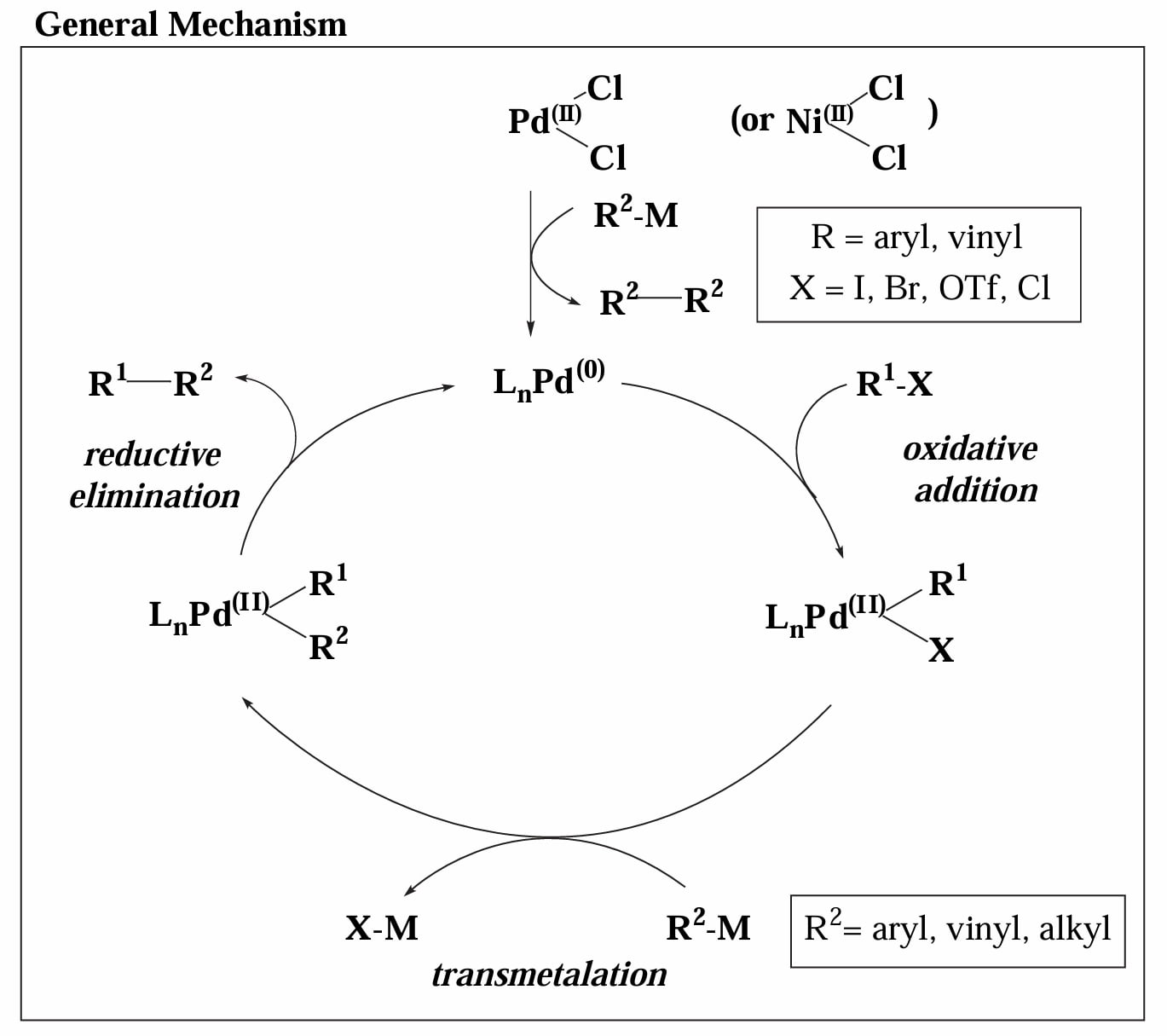
根据在转金属一步中使用的主族金属的不同,我们可以列举下面几个典型反应。
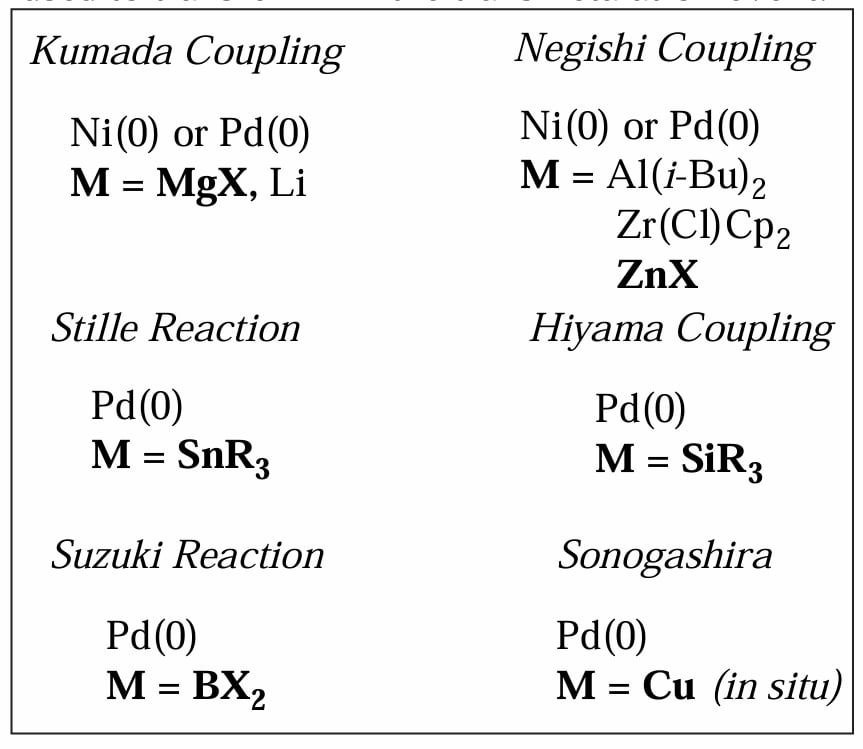
3.2 Kumada coupling
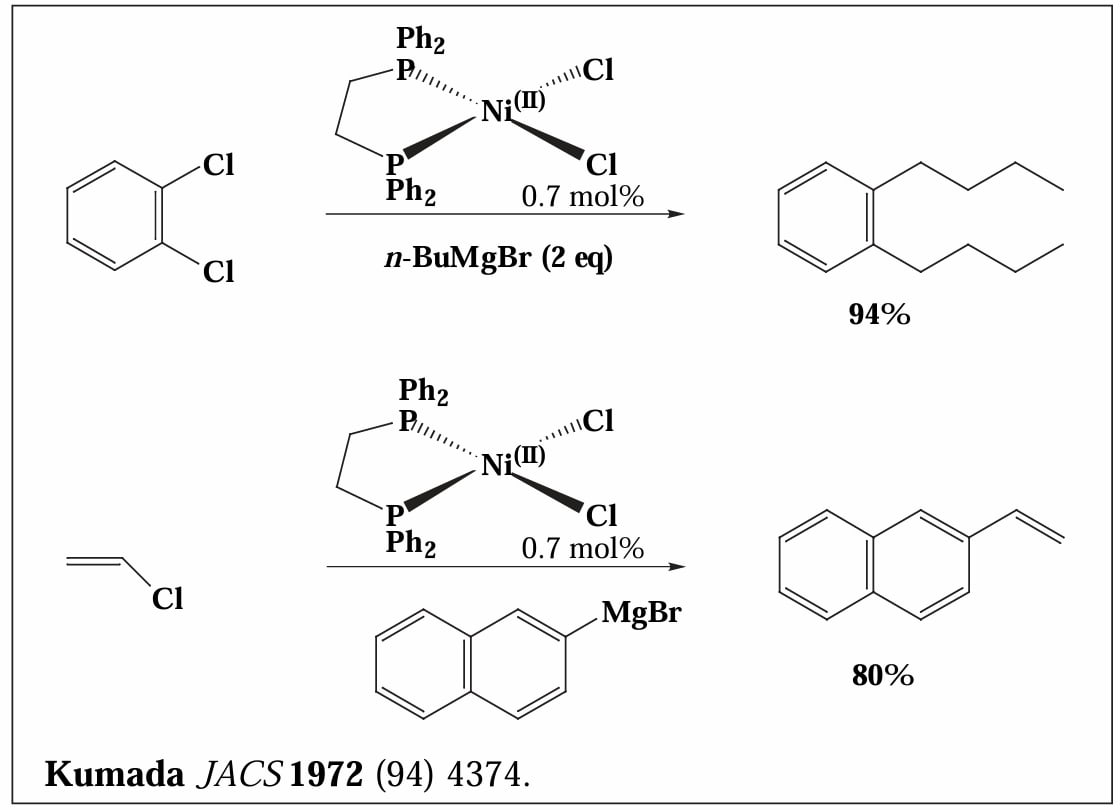
1972年,Kumada 等人报道了 Ni 催化的偶联反应,使用格氏试剂作为转金属试剂。
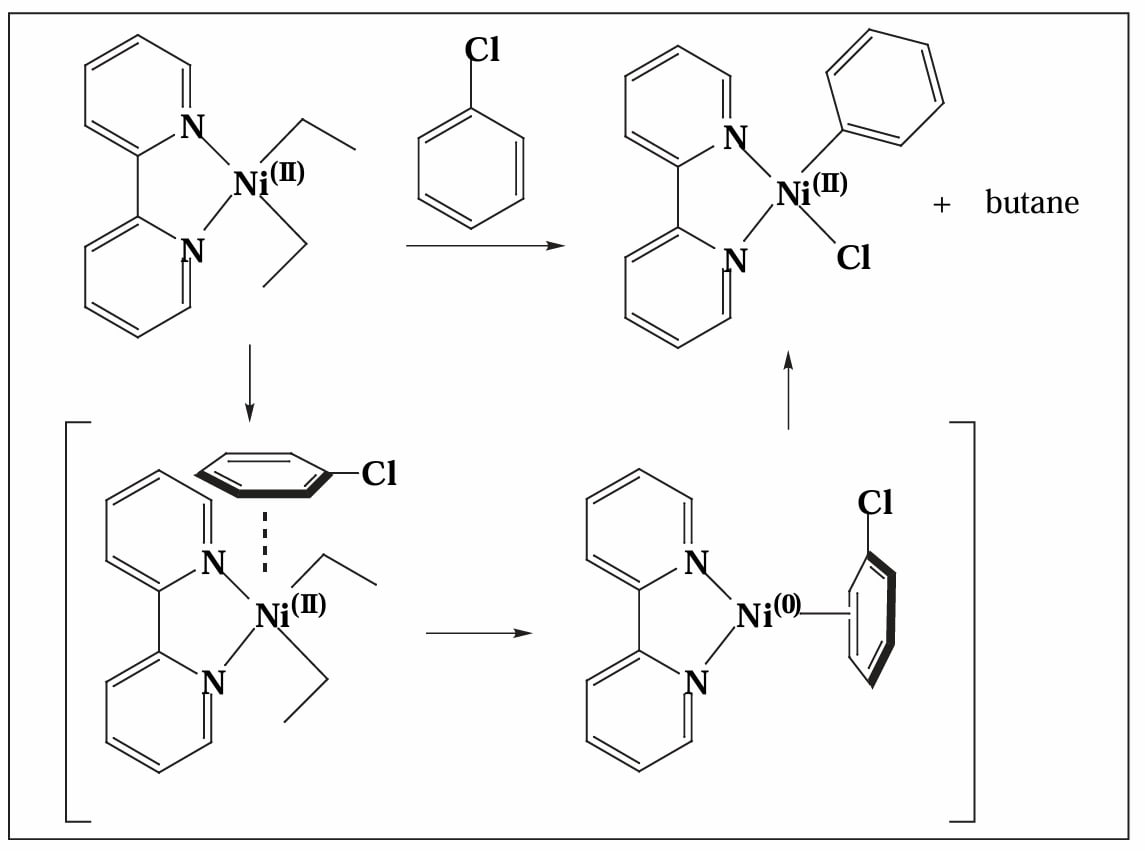
反应使用 Ni(II) 盐作为催化剂,但是真正的催化物种是 Ni(0) 物种,因此第一步需要先生成 。Ni(II) 盐通过两次转金属过程、再还原消除,脱去一个烷基偶联产物,得到 Ni(0)。
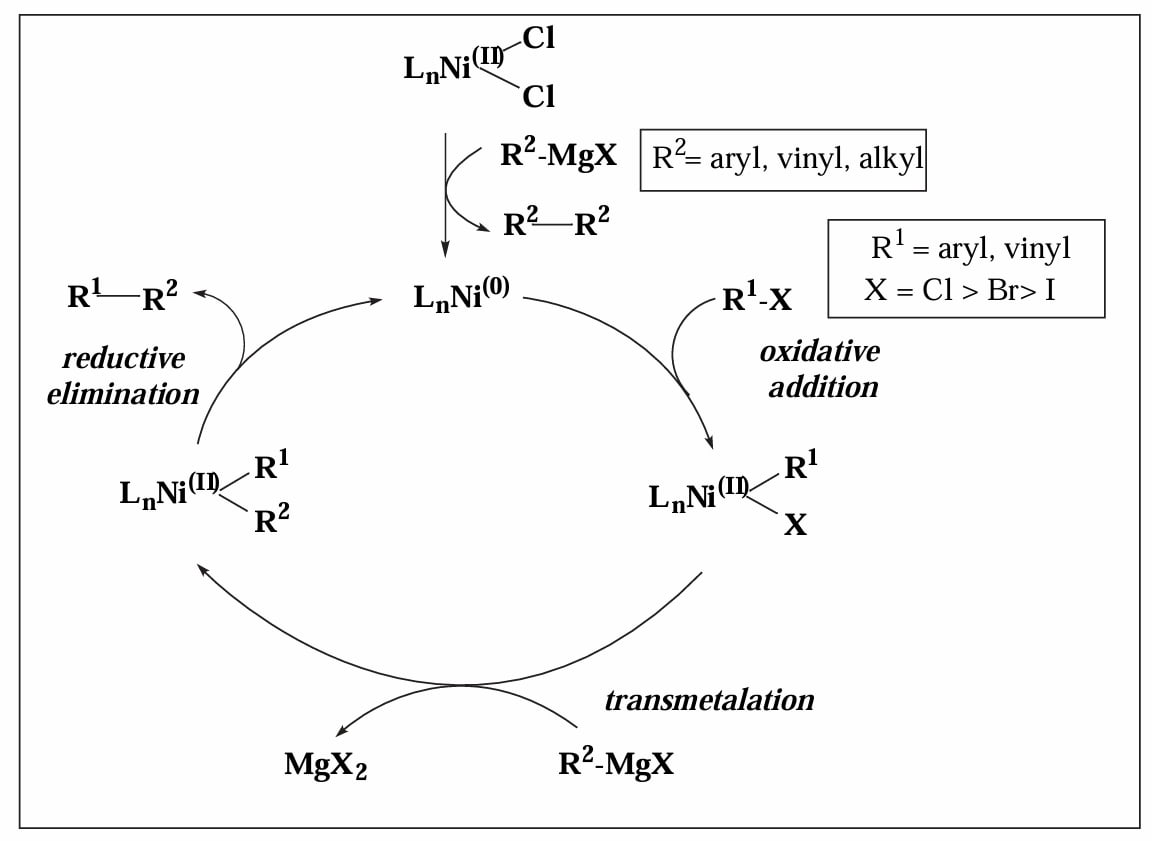
Yamamoto 等人指出,在催化物种生成过程中,芳基卤化物可能作为 π-acid 促进催化剂的还原消除过程,并随后发生氧化加成[1]。
Kumada 反应催化剂的配体常用双齿膦配体(如 dppe/bis(diphenylphosphino)ethane、dmpe/bis(dimethylphosphino)ethane、dmpf/bis(dimethylphosphino)ferrocene 等),其往往表现出比单齿膦配体更好的催化活性。
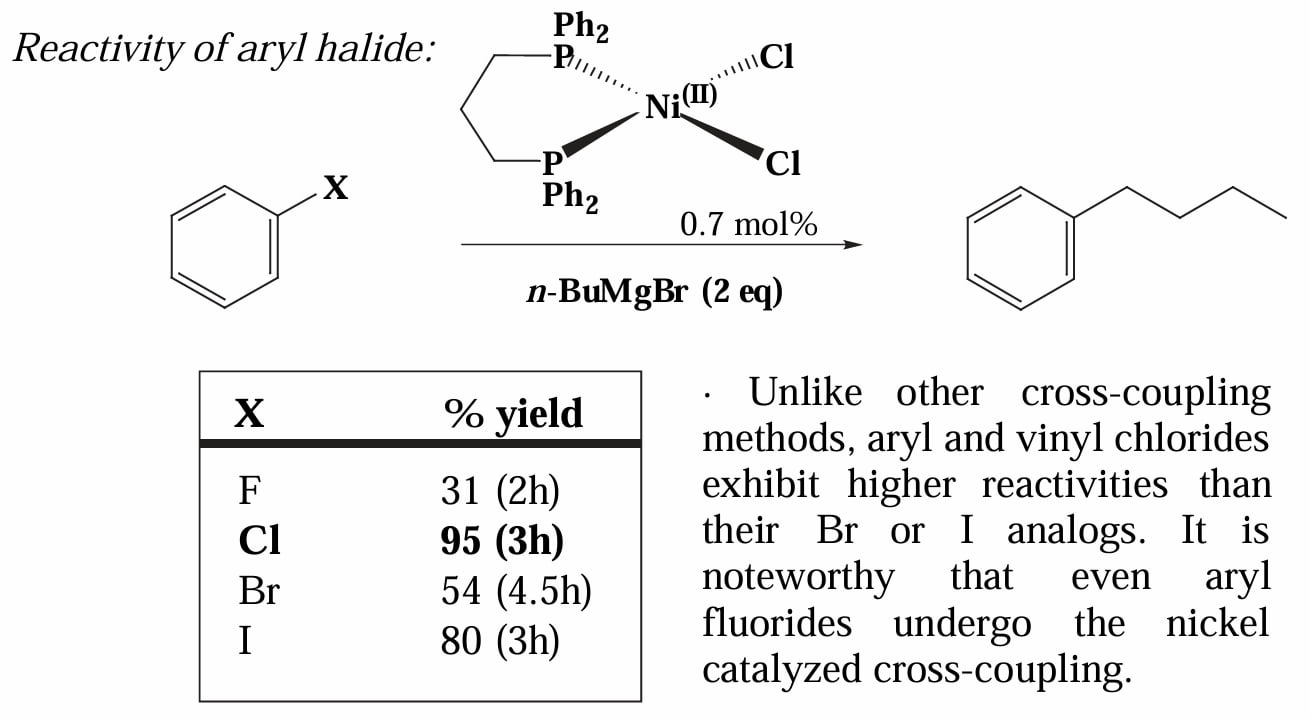
Kumada coupling 在反应性上有独特之处,对于芳基卤化物,其活性表现为 ,且对于芳基氟化物也能进行反应[2]。
Ni 催化的 Kumada coupling 对于烯基卤化物的偶联具有立体专一性,然而对于烯基格氏试剂则无立体选择性。
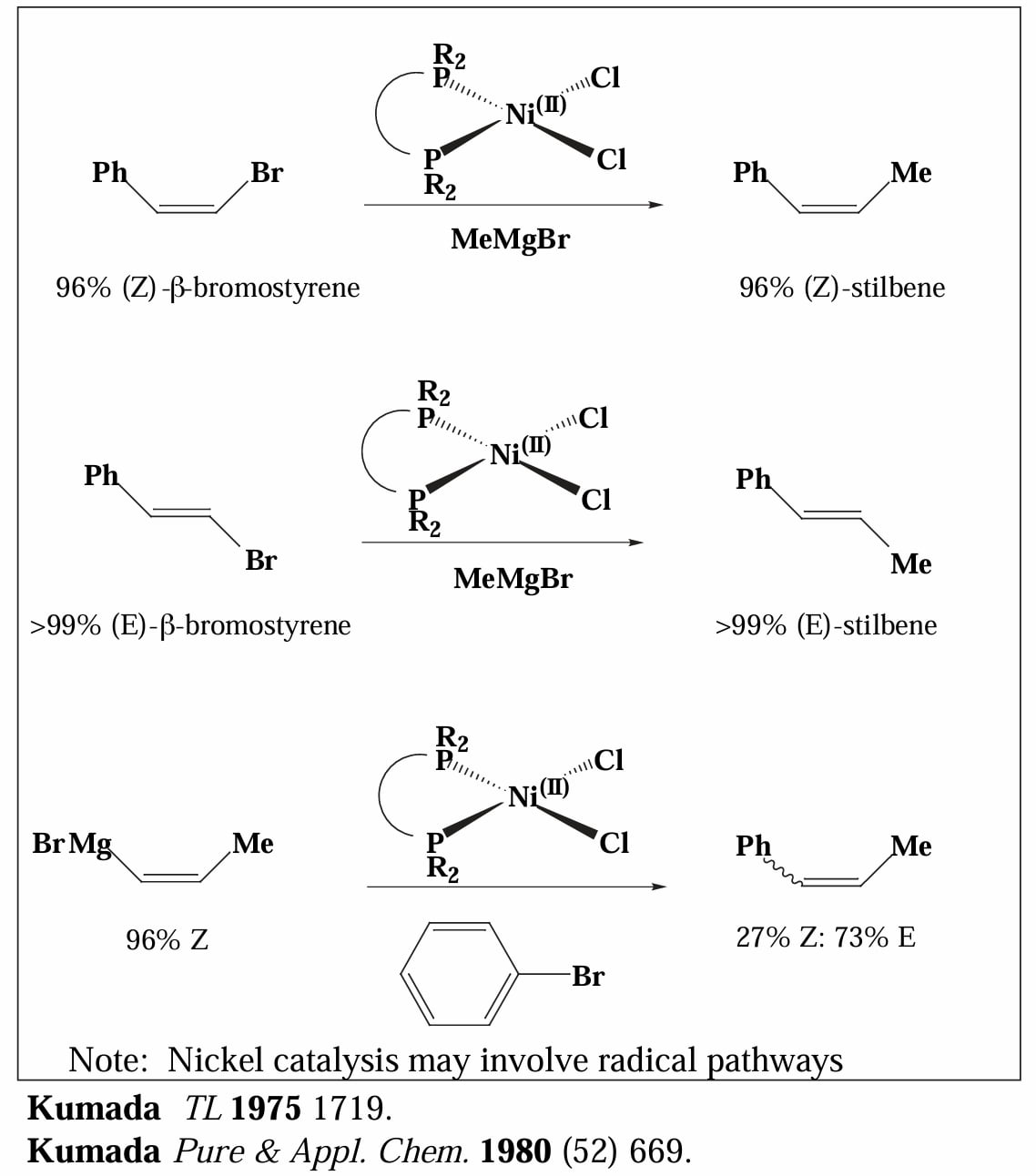
Pd 也可以用于催化 Kumada coupling,且对于烯基卤化物和烯基格氏试剂都具有立体专一性。
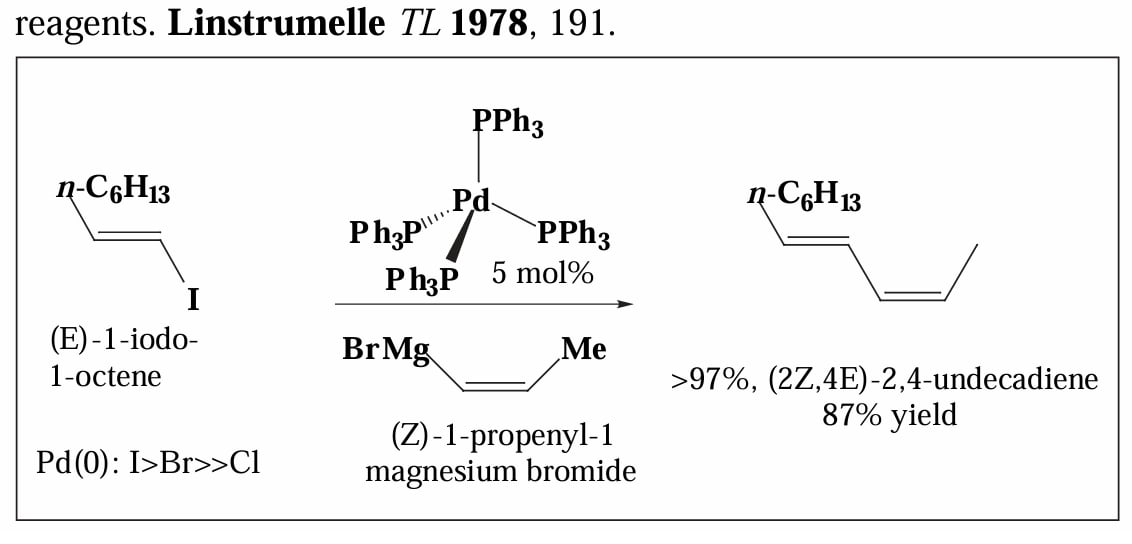
最后看几个应用上的例子。
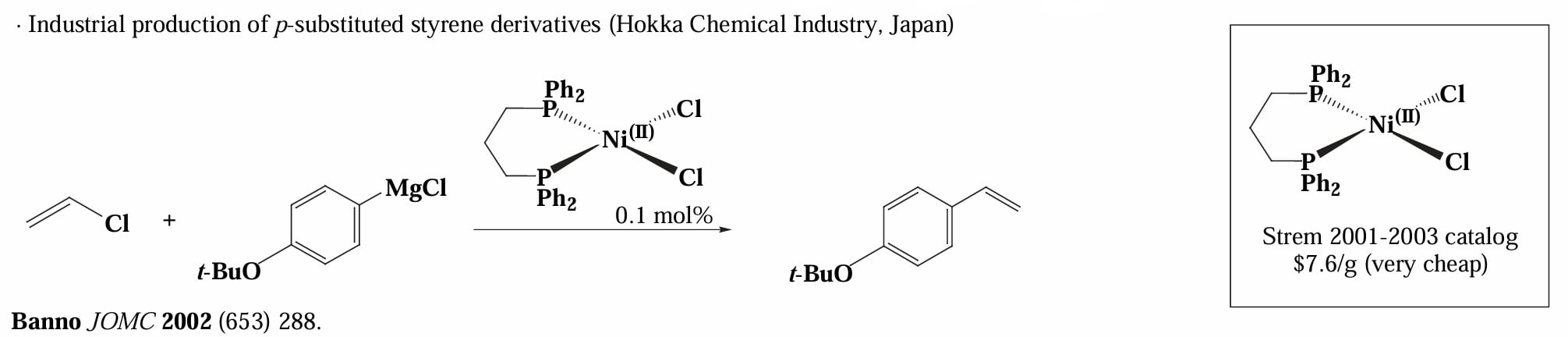
Ni 催化的 Kumada 反应由于催化剂价格低廉,因此被用于工业上大量生产对位取代的苯乙烯衍生物。
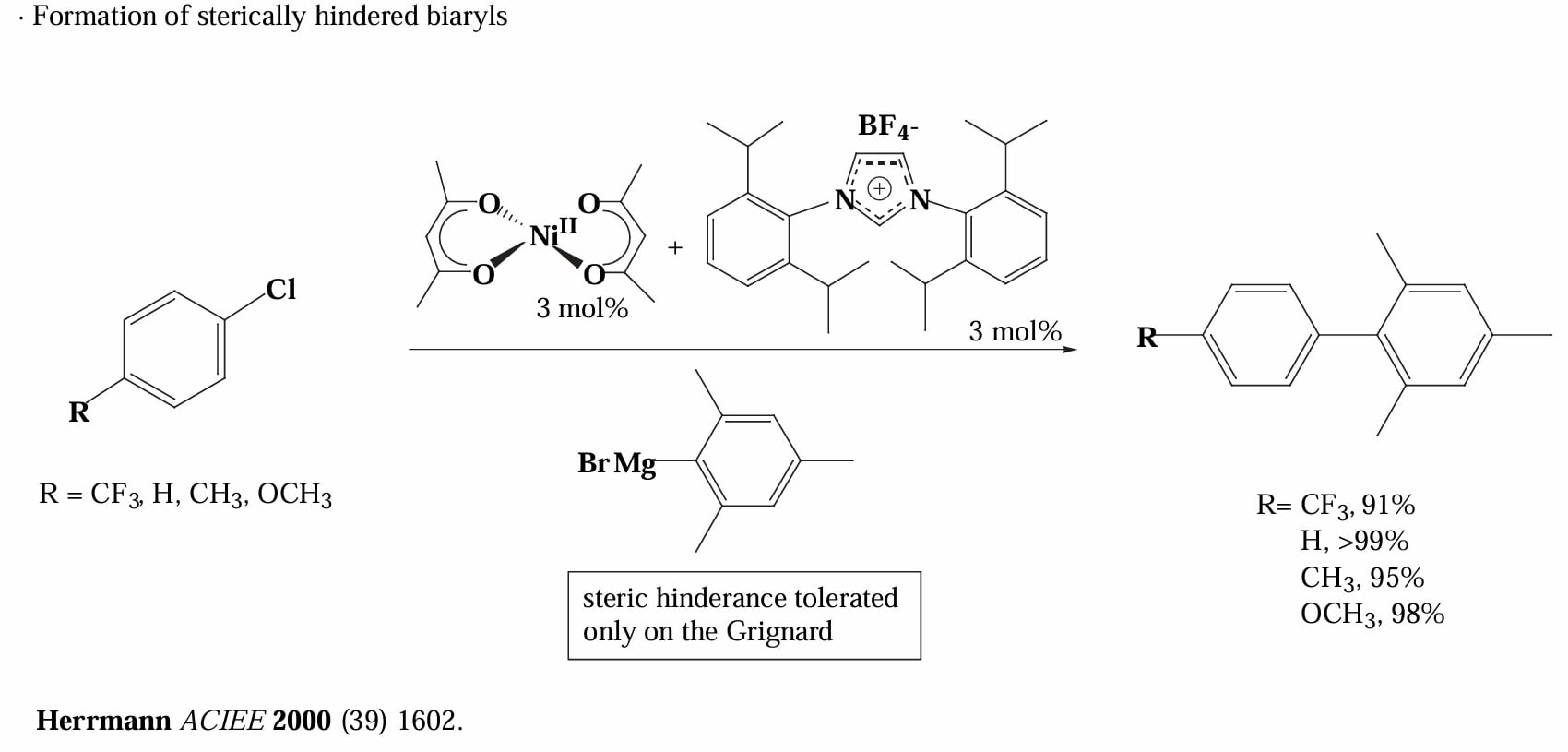
Herrmann 等人报道过使用 Ni 催化剂和 NHC 配体催化的二芳基偶联反应,其对于大位阻的芳基格氏试剂也有很好的产率。
3.3 Negishi Coupling
1980年,Negishi 等人报道了使用 Pd(0) 催化,使用 alkylzinc 作为转金属试剂的偶联反应。
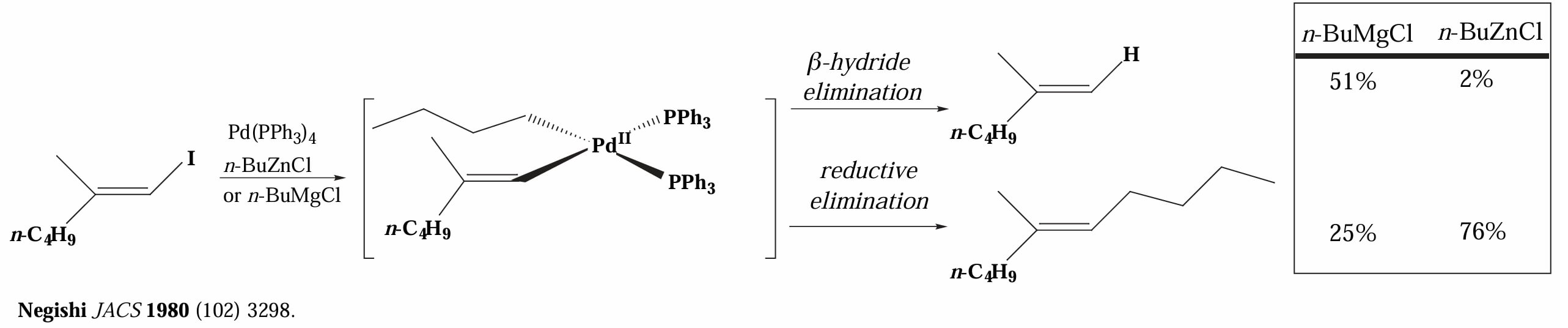
在上面的例子中,使用烷基锌作为转金属试剂得到了更多的还原消除产物,这可能是因为烷基锌作为一个相较格氏试剂更强的 Lewis acid,和 Pd(II) 中间体有更强的 π-acid 效应,使得中心金属更缺电子,因而更容易发生还原消除。
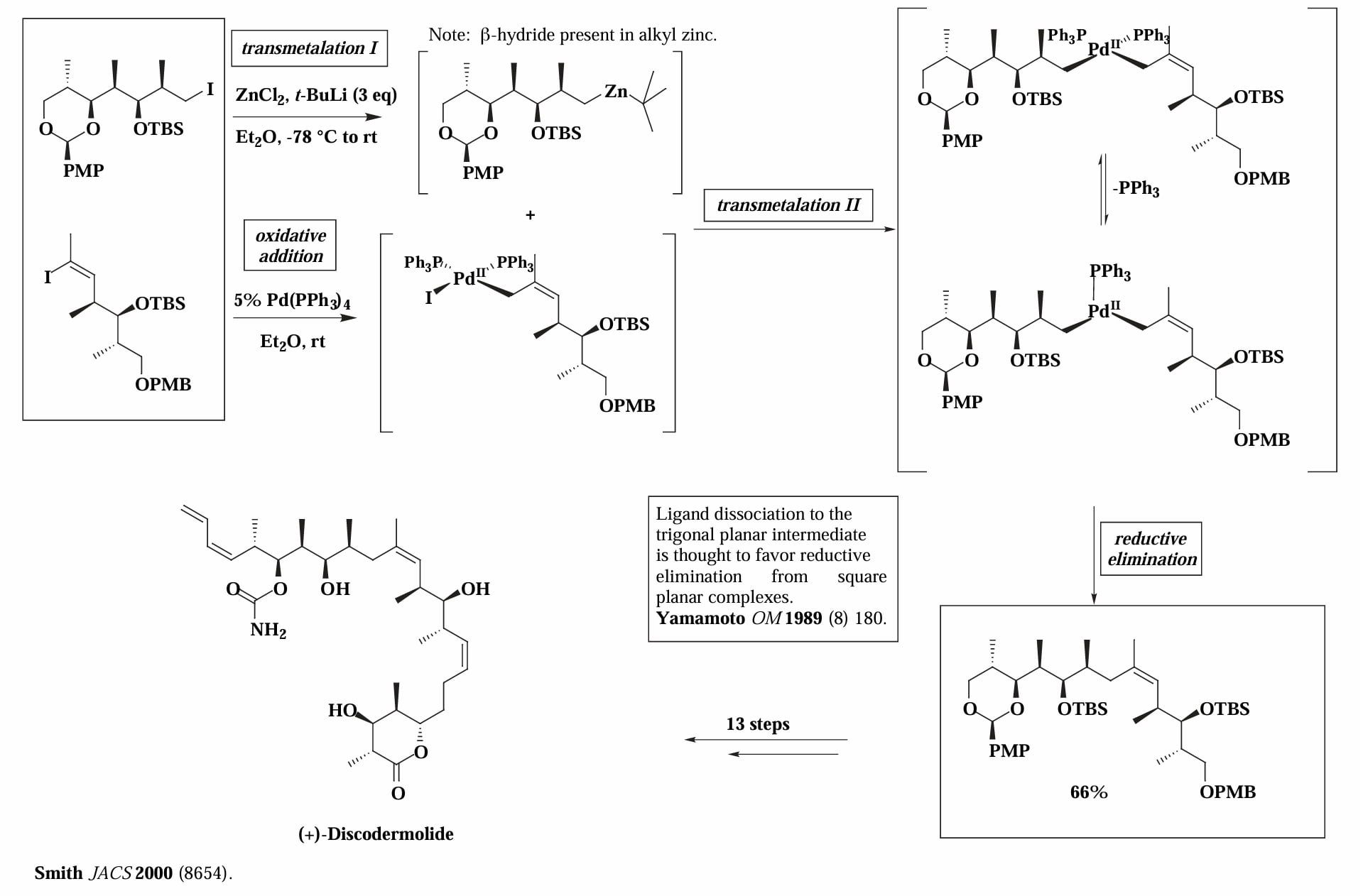
Smith 等人在合成 (+)-Discodermolide 的过程中使用了 Negishi coupling 连接两个片段。
3.4 Stille Coupling
Stille 等人在1979年报道了利用 Sn 试剂作为转金属试剂的偶联反应。
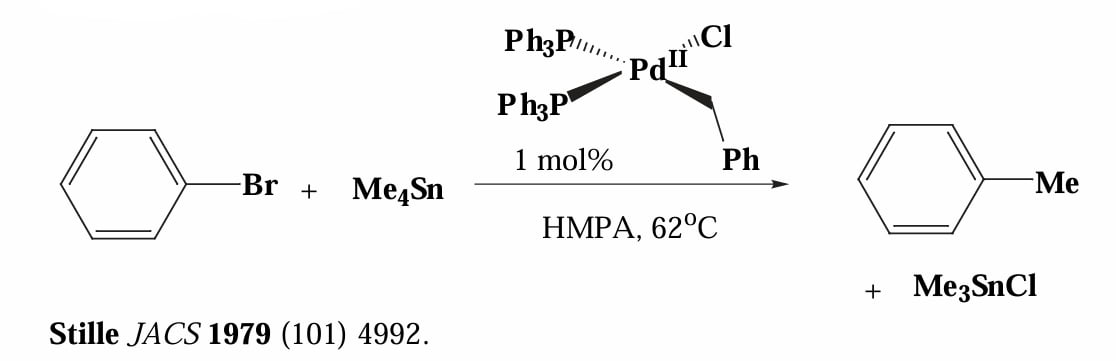
Stille 偶联反应的决速步为转金属,锡试剂的转移速率为炔基 > 烯基 > 芳基 > 苄基 > 烯丙基 > 烷基,常用甲基或者正丁基作为惰性基团,例如使用 来转移 R 基团。
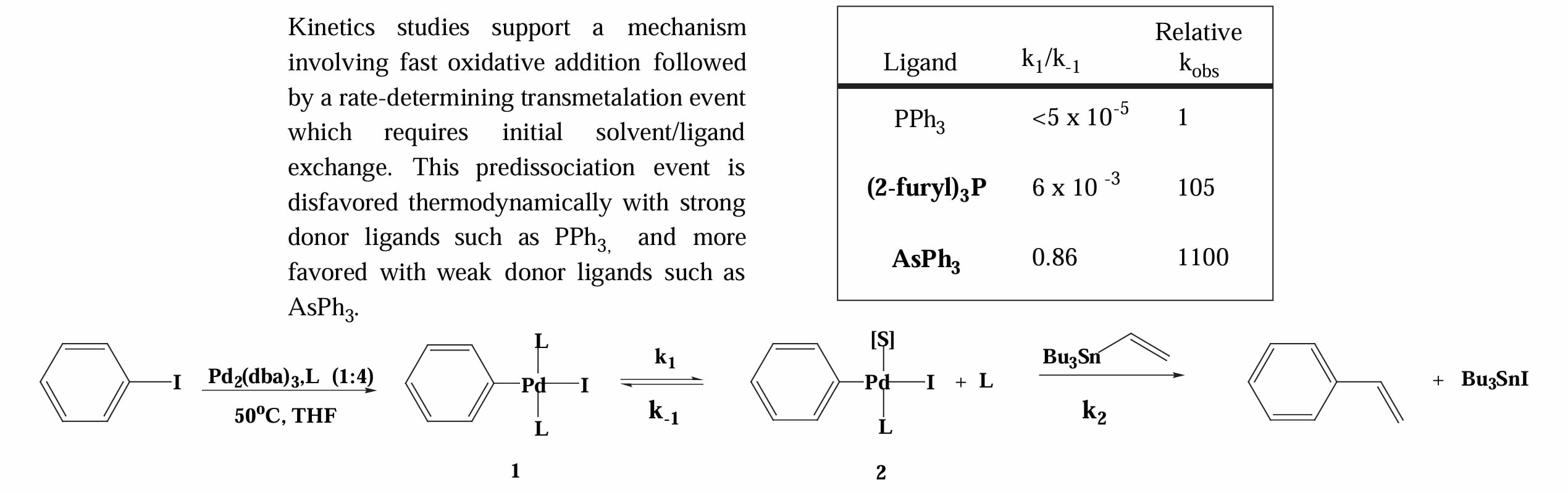
对于氧化加成一步,其插入 C-X 键的速率为 。
Stille 偶联的优点在于对各种官能团的耐受性非常好,且反应条件非常温和。这使得其被应用于各种合成反应中[3]。
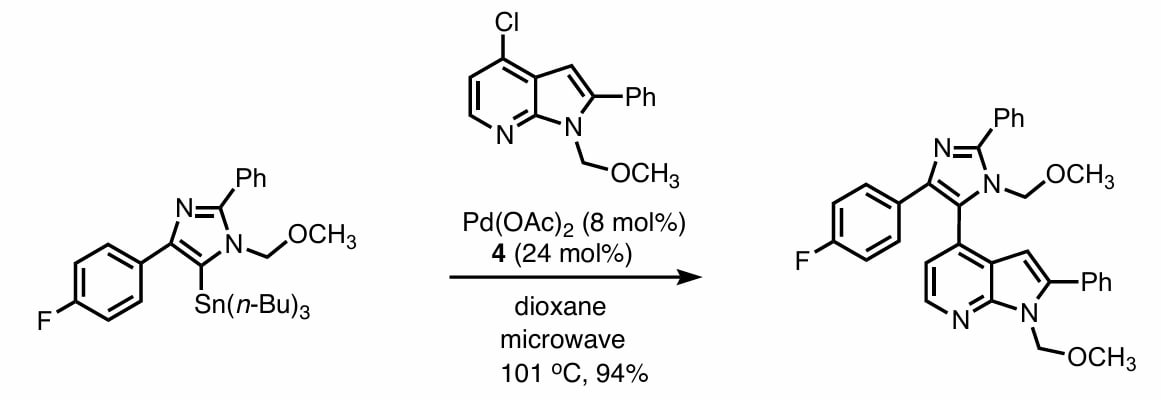
Espinet 等人的机理实验指出[4]:stille 偶联中转金属作为决速步,使用更弱的 donor ligand 更容易在进行转金属时配体更快的离去,从而加速反应。
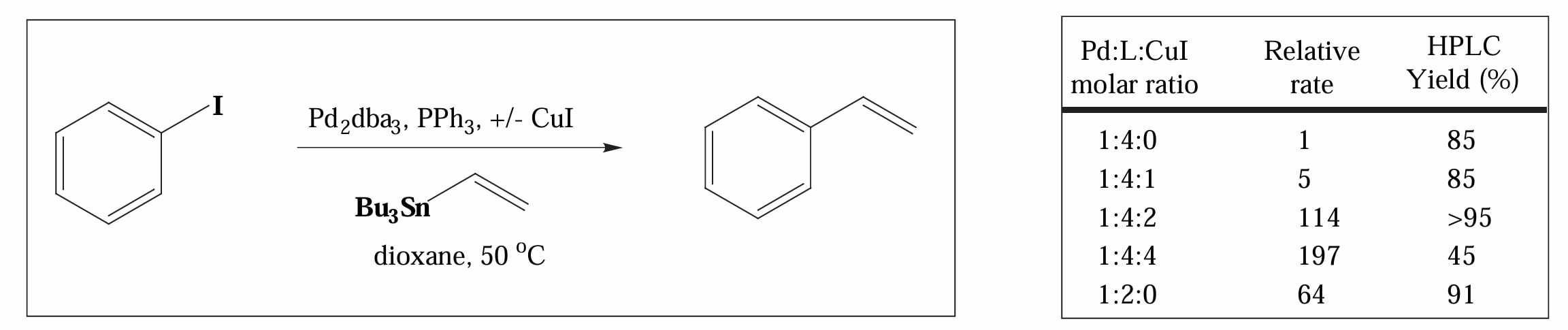
1994年,Farina 和 Liebeskind 等人对 Stille 偶联中存在的 “copper effect”,即在 Stille 偶联反应中加入 CuI 会大大加速反应的现象展开了研究[5]。
他们的实验指出 “copper effect” 的存在原因是 1)CuI 会与游离的膦配体结合,因此有利于在转金属一步中配体的解离,CuI 起到一个 “ligand scavenger” 的作用;2)在大极性溶剂中(如 NMP)CuI 会与锡试剂先发生转金属,生成更活泼的烷基铜化合物,随后再与 Pd 催化剂发生转金属。
Corey 等人在1999年进一步研究发现,对于大位阻的烯基锡底物,使用 CuCl 比 CuI 得到了更高的产率[6],推测可能是因为 CuCl 具有更强的亲电性(Cl 的电负性比 I 更强)。
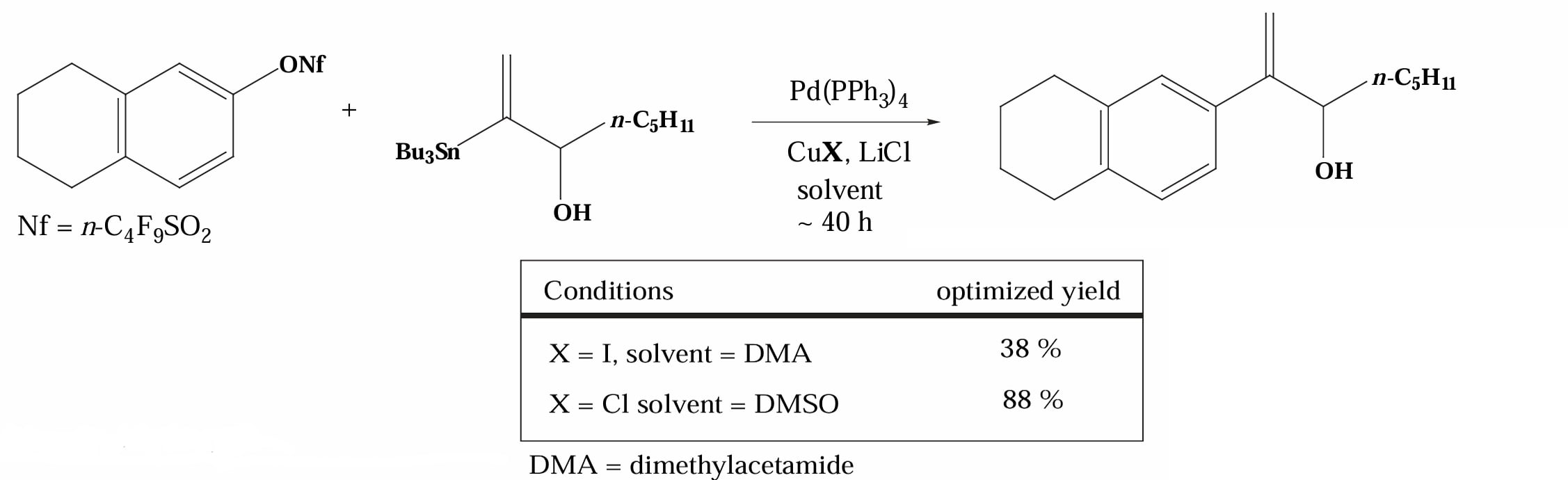
除了在转金属步骤中把转金属试剂转化为更活泼的烷基铜试剂以外,也可以直接提升锡试剂的活性,例如 Lampert 等人在1974年报道了使用 TBAF 作为氟离子源来让单烷基取代的锡试剂更具反应性[7]。原因是 可以配位到 Sn 上从而增强其负电性,易于转移。
Stille 偶联的一个重要特性是反应条件温和,这使得它具有非常好的官能团耐受,被应用于各类复杂合成当中,以下是几个例子。
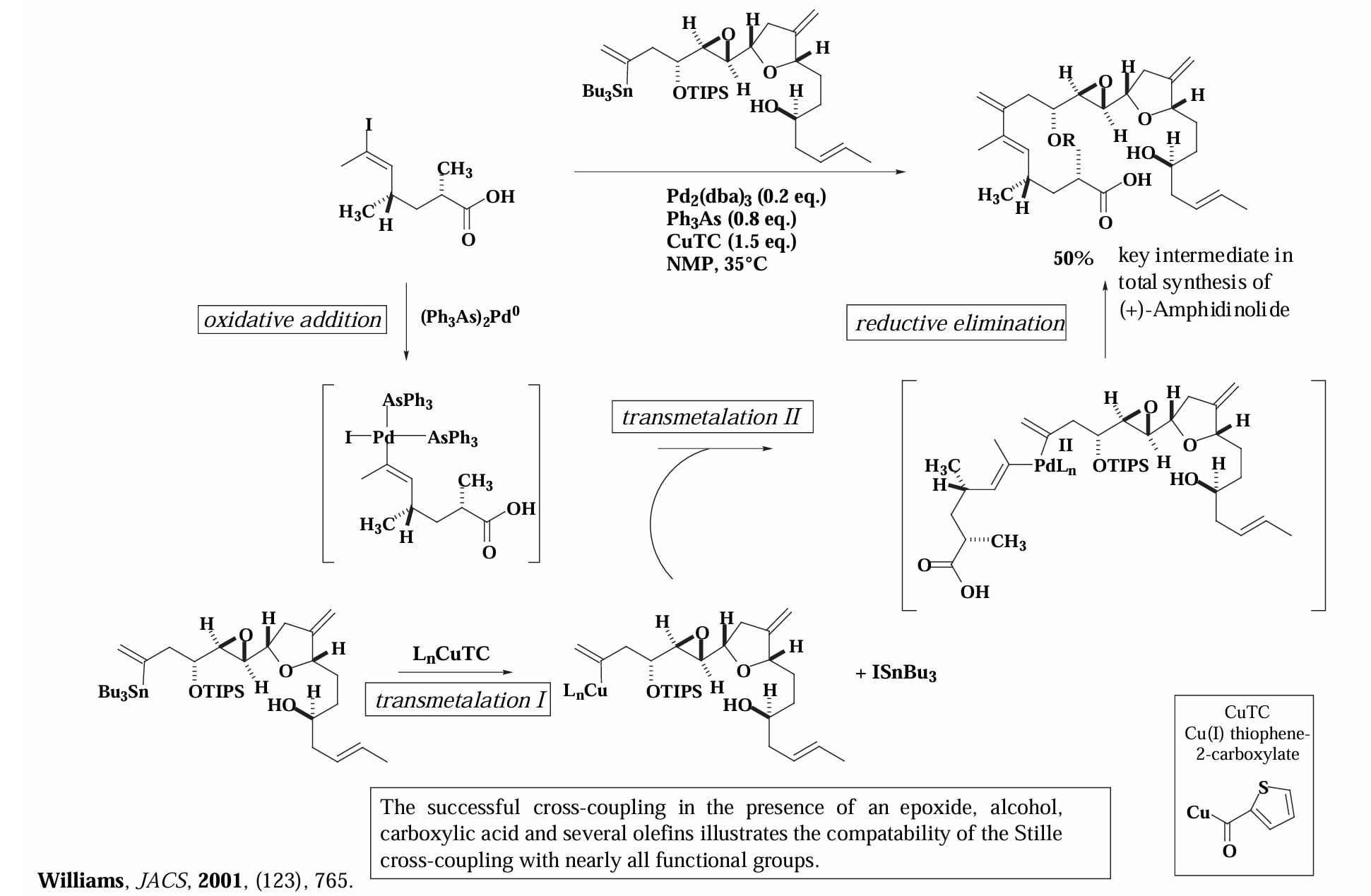
Williams 等人对 (+)-Amphidinolide 的全合成。
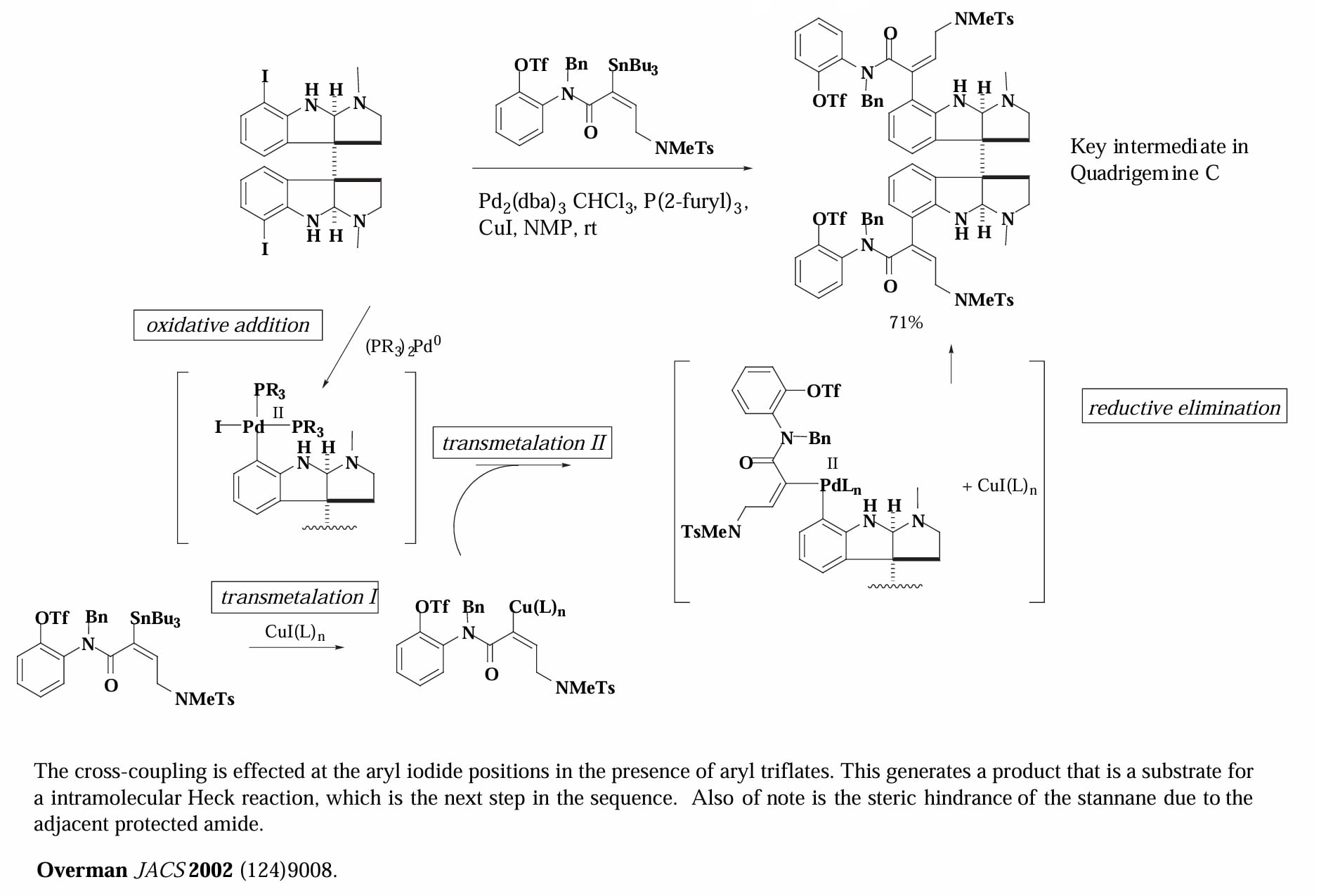
Overman 等人对 Quadrigemine 的全合成。
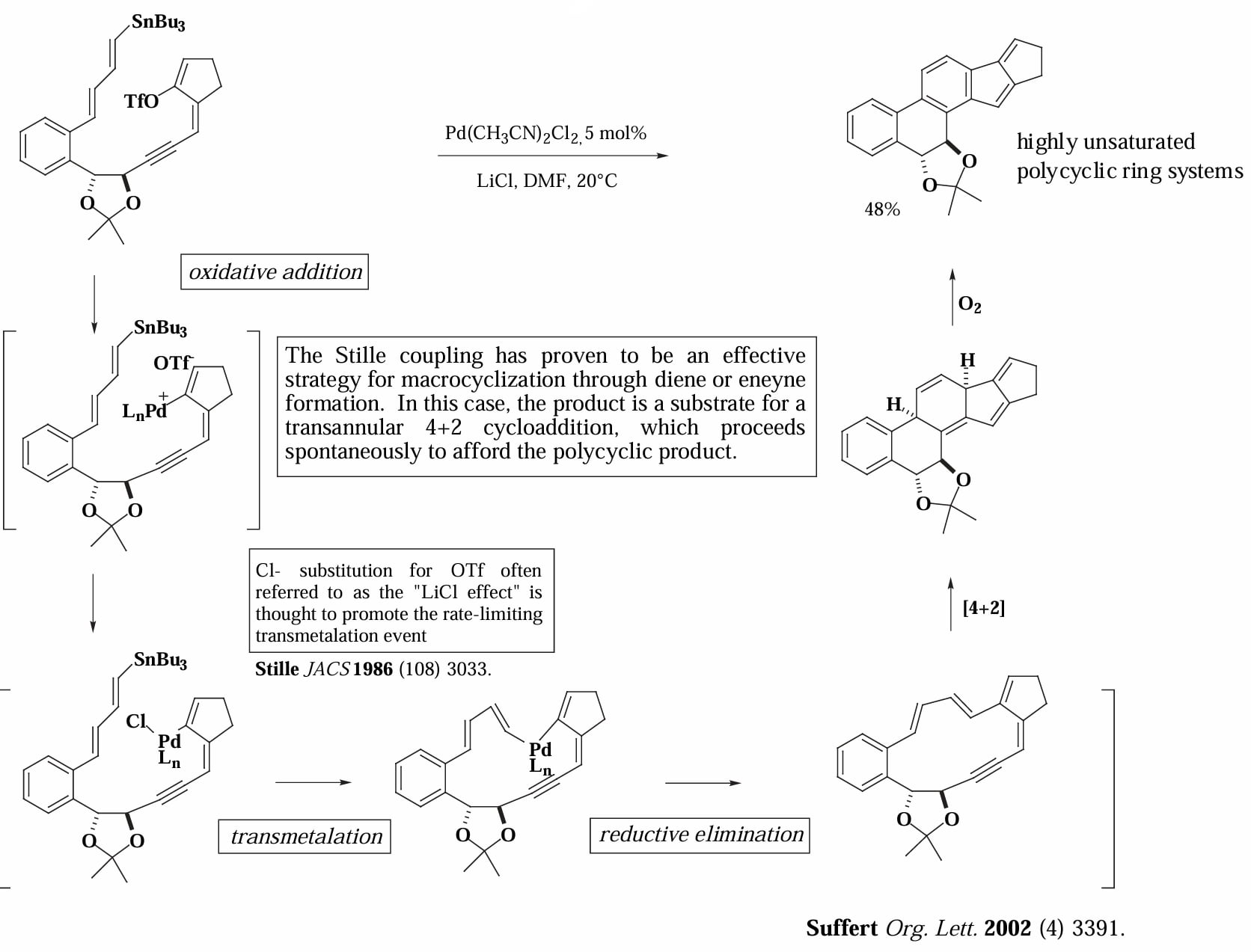
Suffert 等人对大环化合物的合成。
3.5 Hiyama Coupling
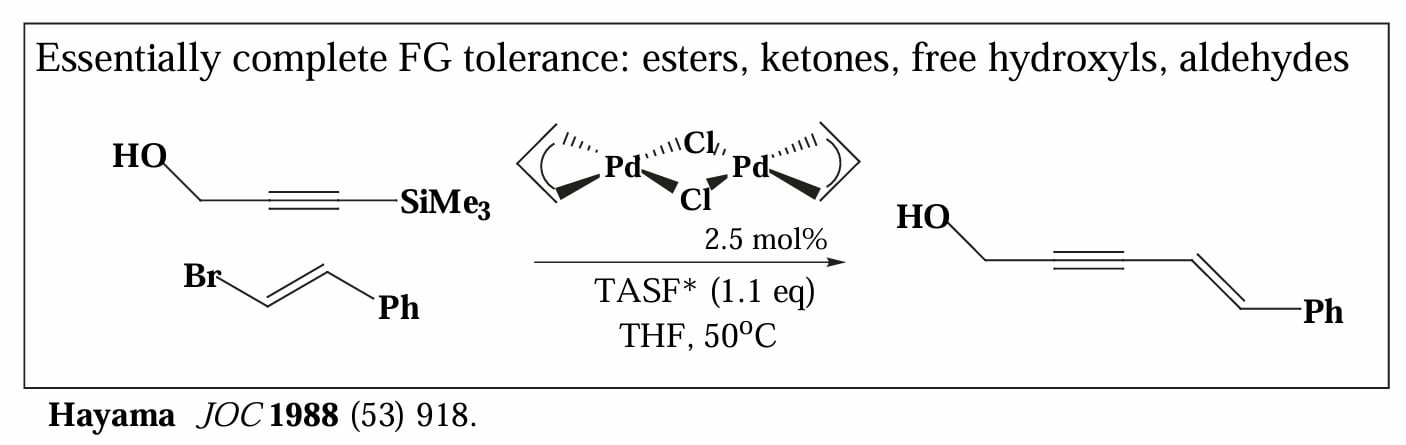
1988年,Hiyama 等人报道了利用有机硅试剂作为转金属试剂的偶联反应,该反应的显著特点是需要提供 源才能顺利进行,体系中的 可以与硅试剂结合,形成五配位的硅化合物,增强其亲核性,有利于发生转金属反应。
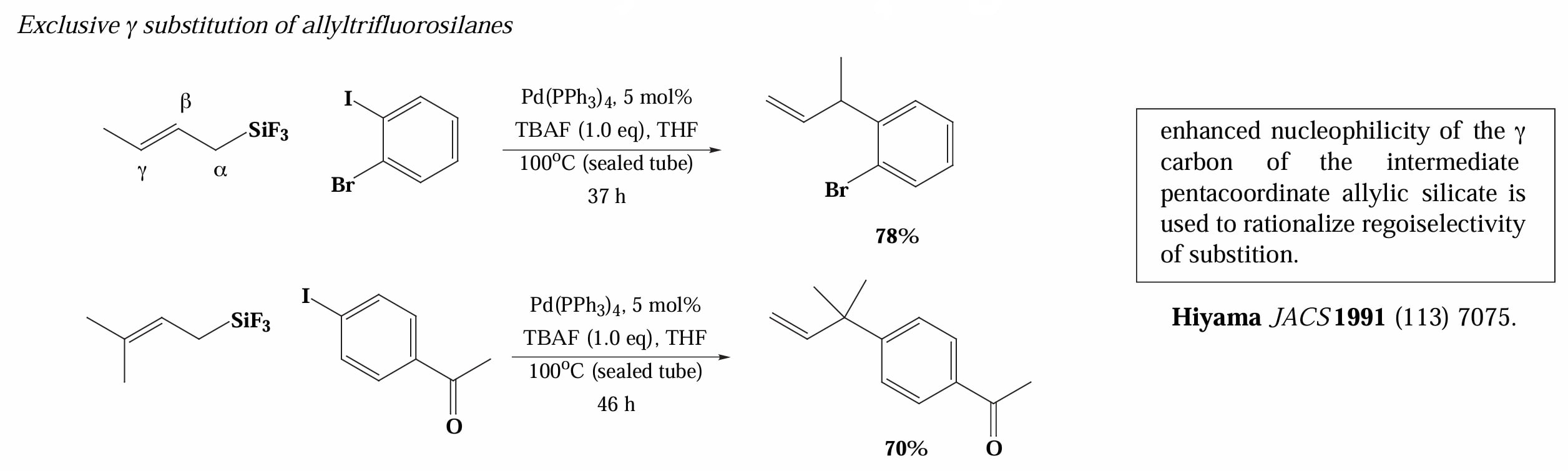
Hiyama 等人还发现烯丙基氟硅烷具有独特的 γ 位反应性,可以高选择性的得到 γ-位取代产物。Ushio 等人通过计算指出其选择性来自于 配位后 γ-位亲核性的增强,推测是因为 C-Si 键与双键的 σ-π conjugation 导致的[8]。
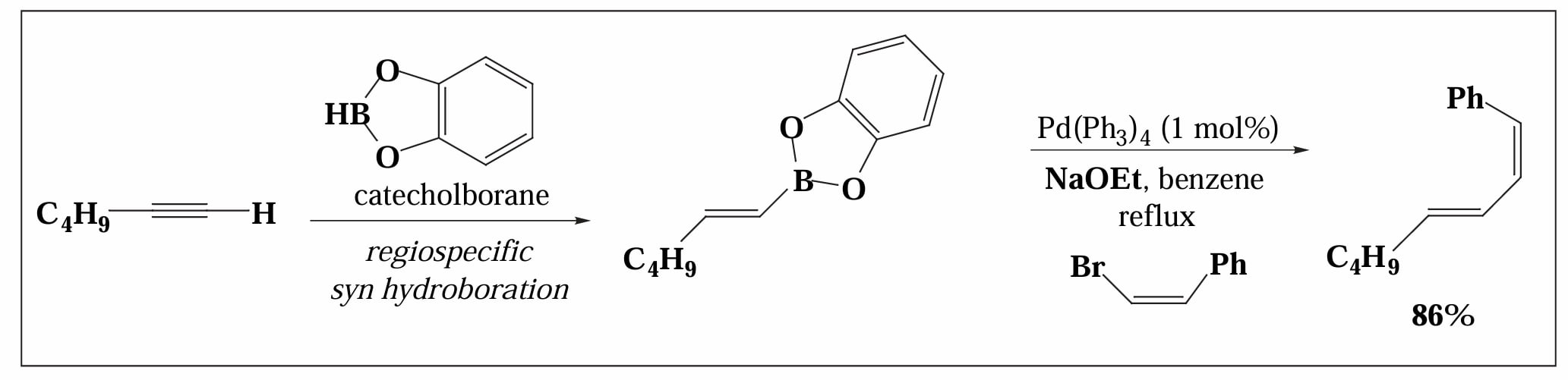
3.6 Suzuki Coupling
1979年,Suzuki 等人报道了利用硼试剂作为转金属试剂的偶联反应[9]。
硼试剂相比锡试剂的一个显著优点是无毒且对空气/水汽稳定,这使得 Suzuki 偶联比 Stille 偶联更安全易行。常用的硼试剂包括 、pinacolborane、9-BBN 等等。
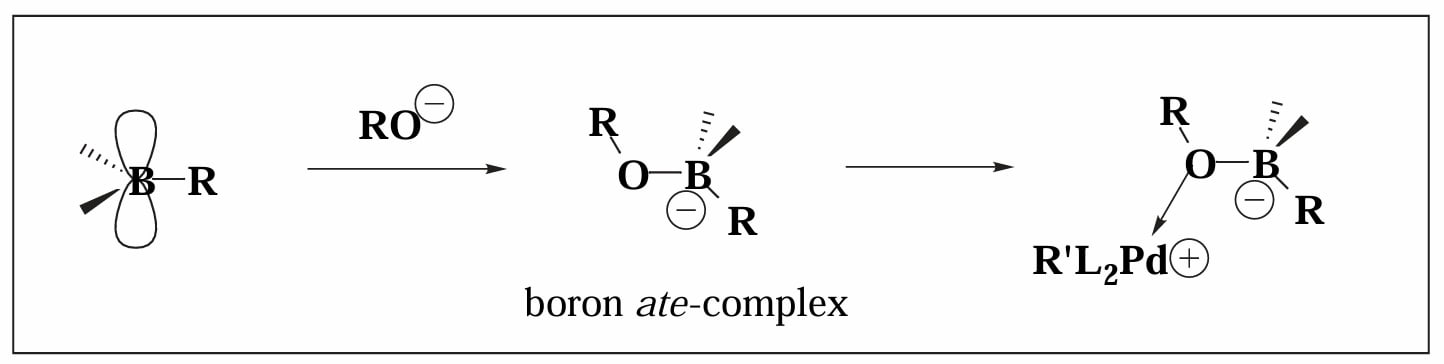
硼试剂对水不敏感的原因是 C-B 键本身具有高度的共价性,但是这样的特性也使得 C-B 键在转金属过程中不易断裂。因此在反应中,我们往往需要外加入碱使得有机硼化合物形成 “ate-complex” 增强 C-B 键的亲核性。
在 Suzuki 偶联的各种偶联底物中,芳基氯是活性最低的物种,其原因来自于 C-Cl 键较高的键能(例如 PhCl B.E.=96 kcal/mol; PhBr B.E.=81 kcal/mol; PhI B.E.=65 kcal/mol)。但是考虑到芳基氯化物便宜易得的特点,对芳基氯化物的直接偶联(即不需要 EWG 的活化)是非常具有吸引力的。
1998年,Fu 等人发现使用大位阻且富电子的膦配体能够克服不利的反应性,实现无活化的芳基氯化物偶联。
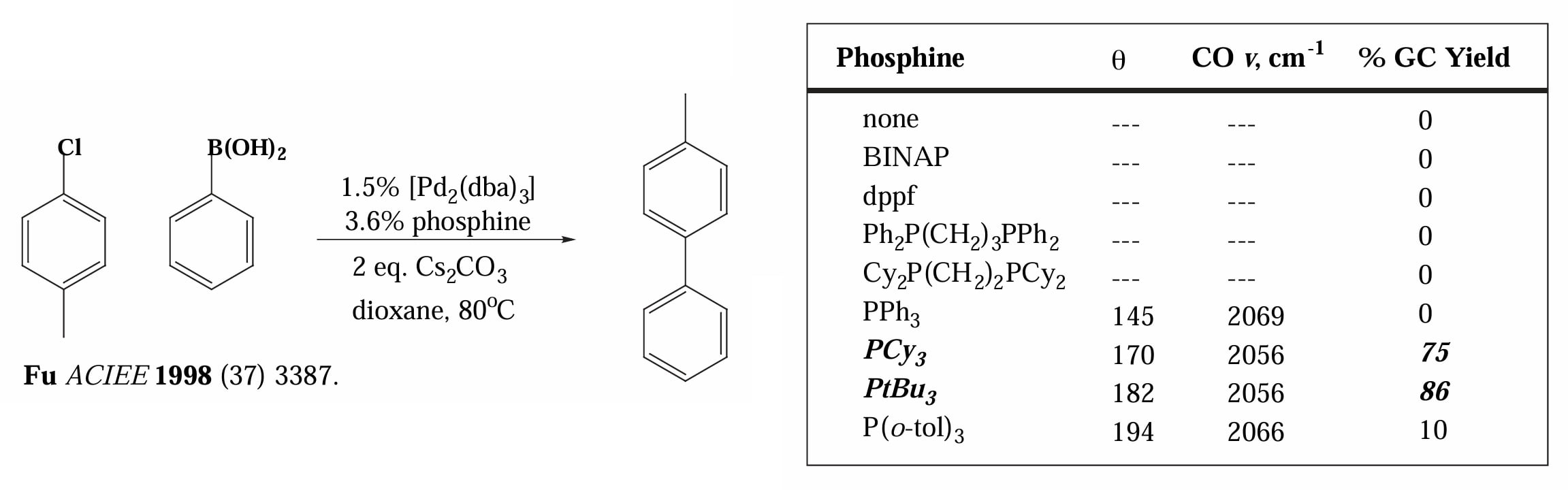
大位阻富电子的配体能够促进氧化加成的可能原因是配体使金属中心更富电子,且大位阻配体会使得金属的配位不饱和,因此易于发生氧化加成增加配位数和降低电子密度。
Fu 等人还发现,利用大位阻富电子配体能够进行一般条件下难以发生的烷基-烷基偶联。
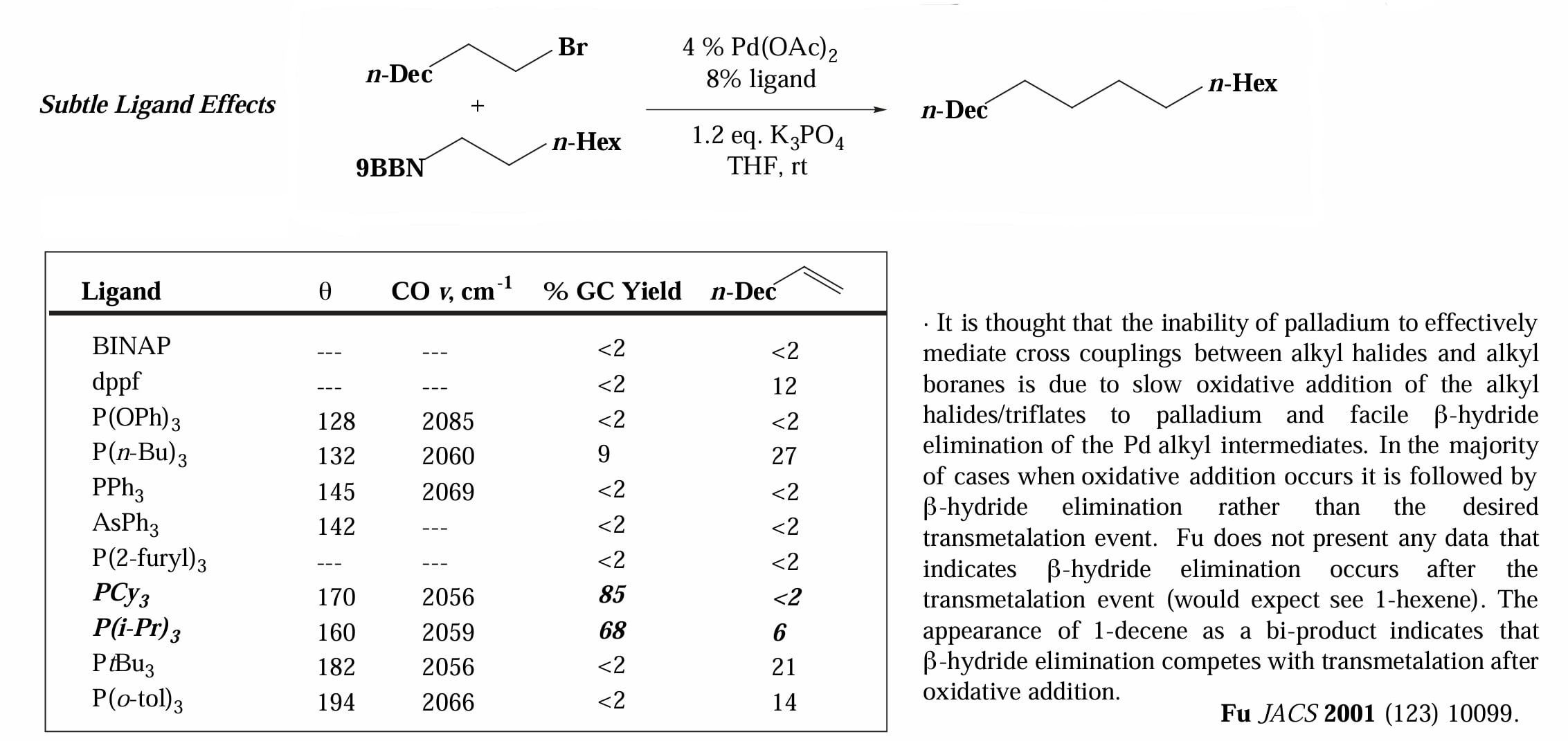
烷基-烷基偶联除了氧化加成难以进行外,另一个不利因素是会发生 β-elimination 的竞争反应,非常好的是,大位阻富电子配体也能够抑制 β-elimination 的发生,因为大位阻配体会让 β-H 难以接近金属,同时给电子能力使得金属中心富电子,因而削弱了 β-H 的 agostic interaction。
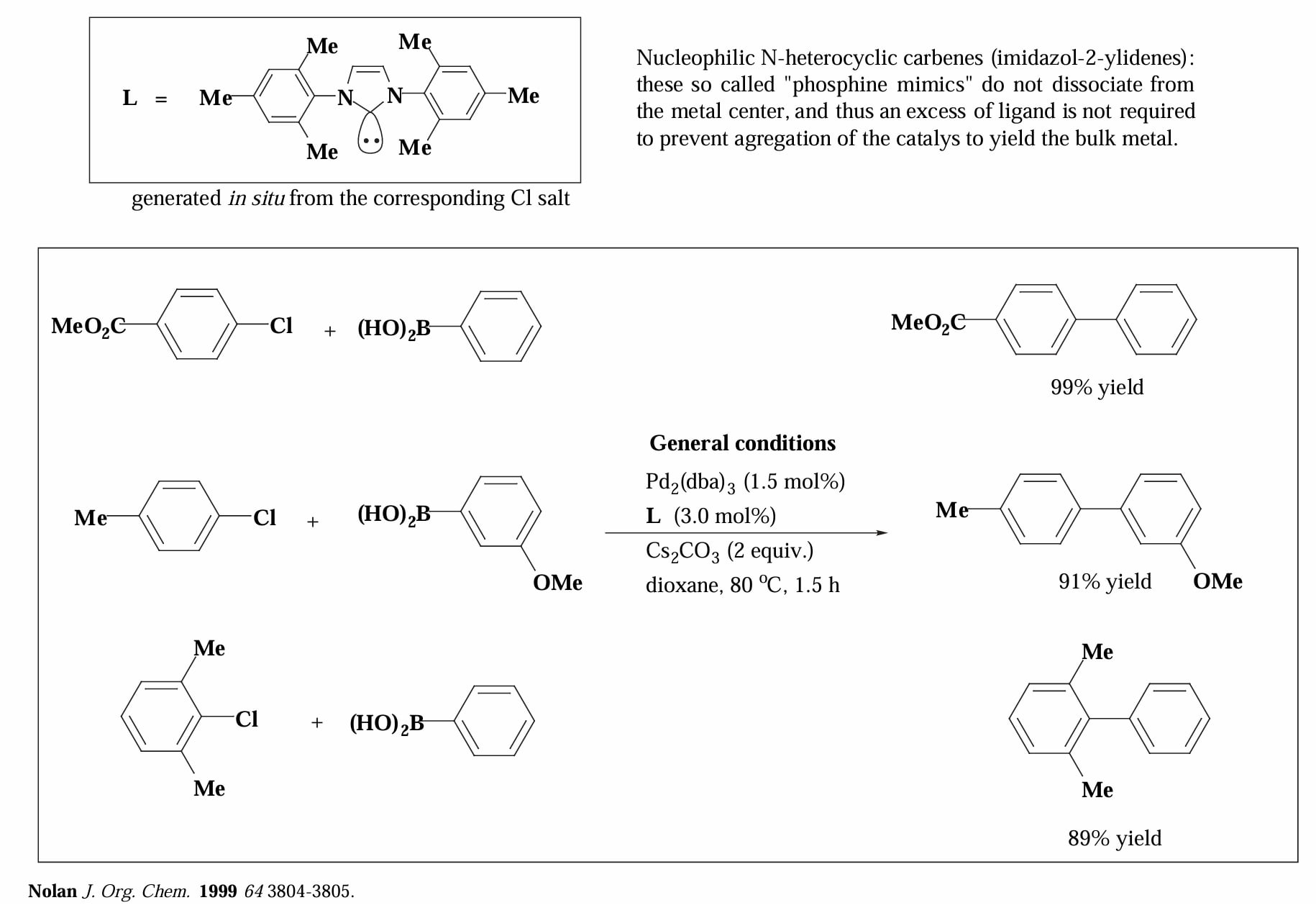
还有一种比较特殊的配体是氮杂环卡宾(N-heterocyclic carbenes, NHC),其特点是与金属结合后不会解离,因此不需要加入过量的配体。
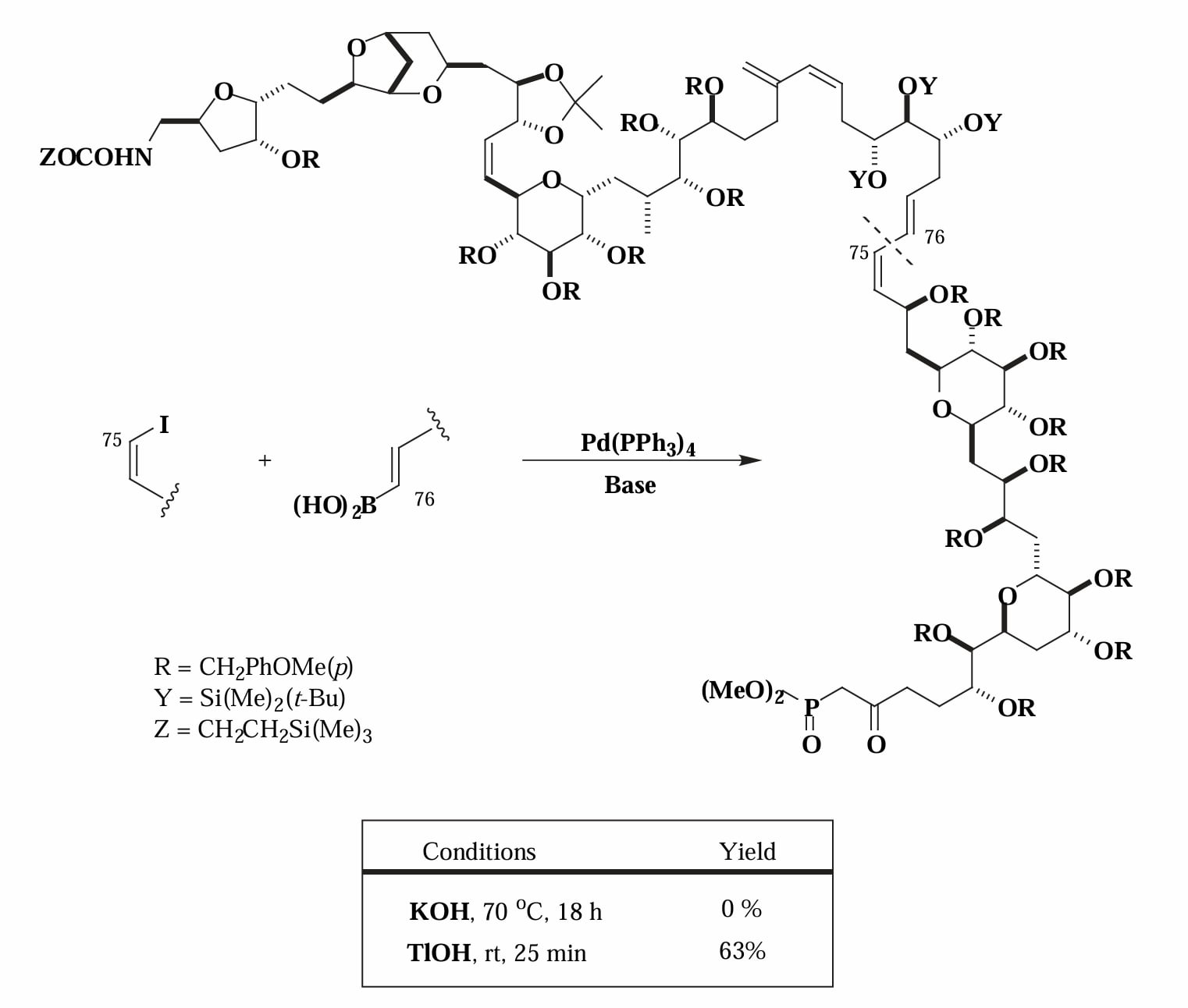
1987年,Kishi 等人发现使用 TlOH 代替常见的 KOH 作为碱会让 Suzuki 偶联反应速率大大增加[10],其反应速率约增加1000倍,反应在0°C下几乎瞬间完成,因此可以适用于大分子量片段的连接,以及含敏感官能团分子片段的连接。Kishi 使用该方法实现了 palytoxin 部分片段的合成。
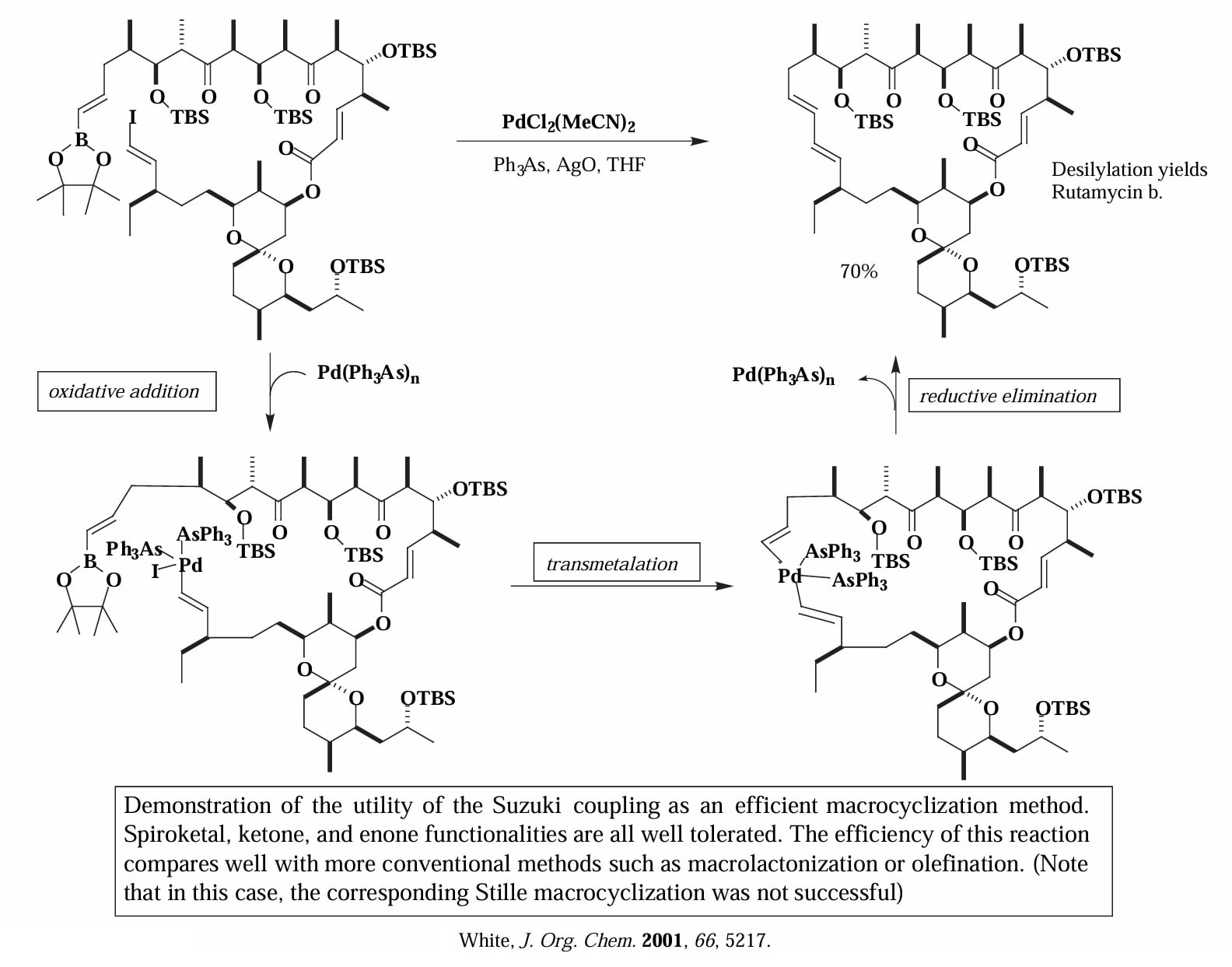
Suzuki 偶联也能够被用于分子的晚期大环关环。
3.7 Sonogashira Coupling
1975年,Sonogashira 等人报道了 CuI 催化的端炔原位去质子化,随后与烯基溴发生偶联反应。
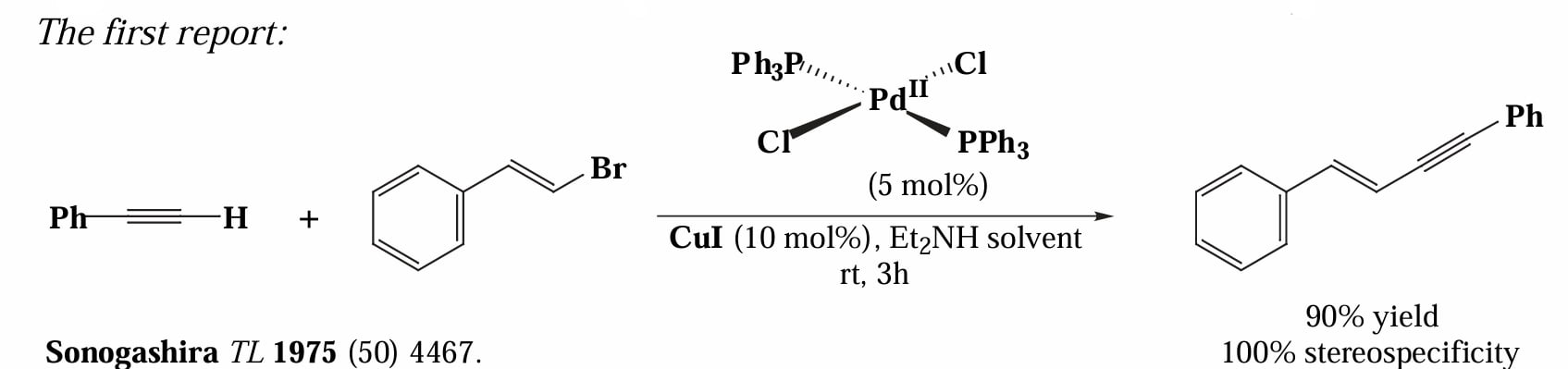
该反应中 底物的反应性是烯基碘/烯基溴 > 芳基碘 > 烯基氯 >> 芳基溴,从这个顺序可以看出,总体上烯基卤化物的反应性要大于芳基卤化物。
Buchwald 和 Fu 等人报道了使用 作为配体实现的芳基溴化合物和炔基的偶联,此处大位阻的 表现出独特的反应性,其他的膦配体都无法顺利催化该反应进行。

Sonogashira 偶联对酯基、羟基、烯丙基醚等基团都有很好的耐受性,因此是完成 偶联的很好的选择。
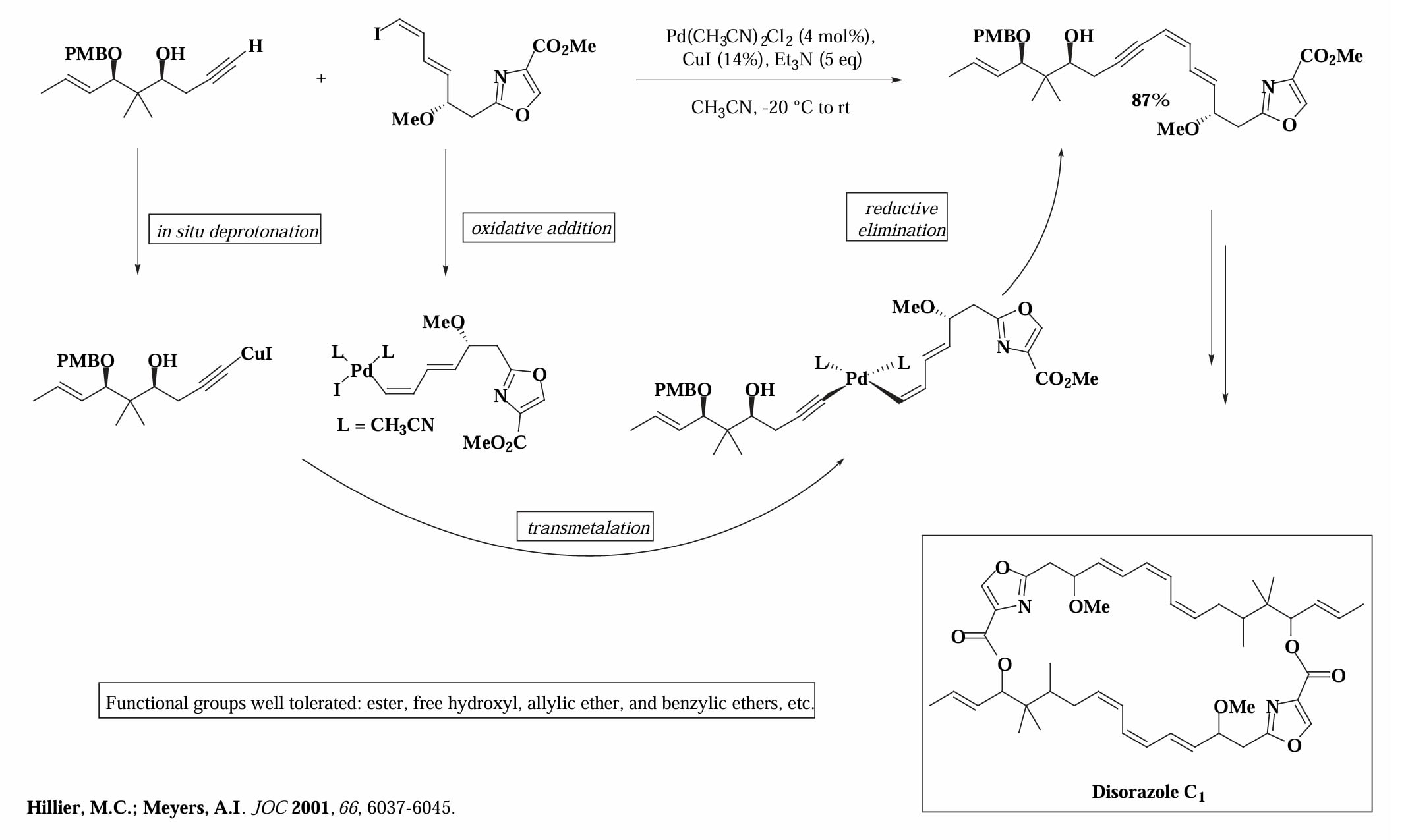
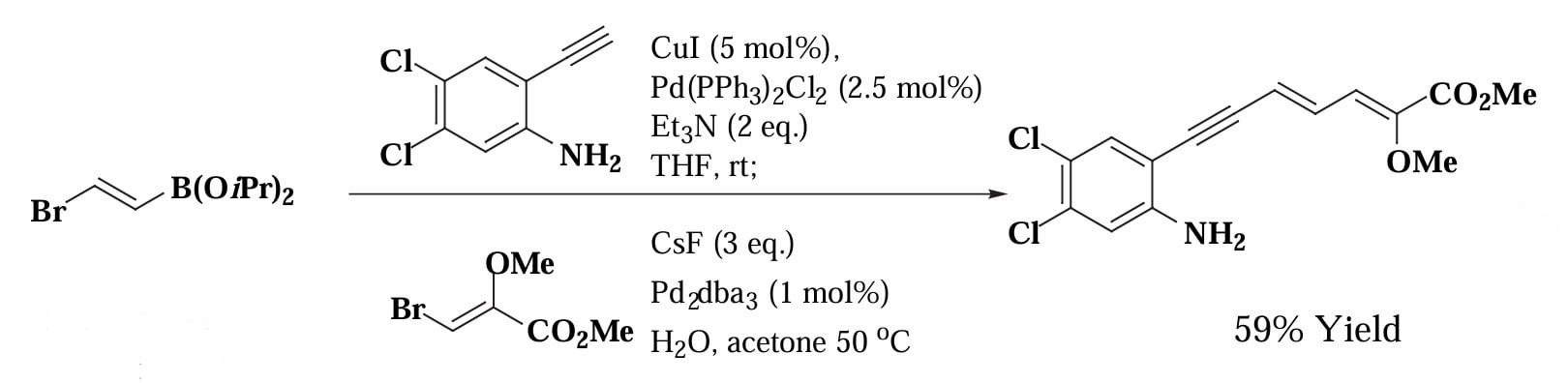
把 Sonogashira 偶联和 Suzuki 偶联串联起来使用,可以快速构建多个共轭烯烃的复杂产物[11],其反应的关键是烯基上同时连有溴和硼烷的底物,可以分别发生不同的偶联,在两端连接两个片段。
3.8 Buchwald-Hartwig Amination
1983年,Migita 报道了通过 Stille 偶联完成的胺化反应,他借助 Sn-N 的转金属化把芳基溴和胺连接起来。
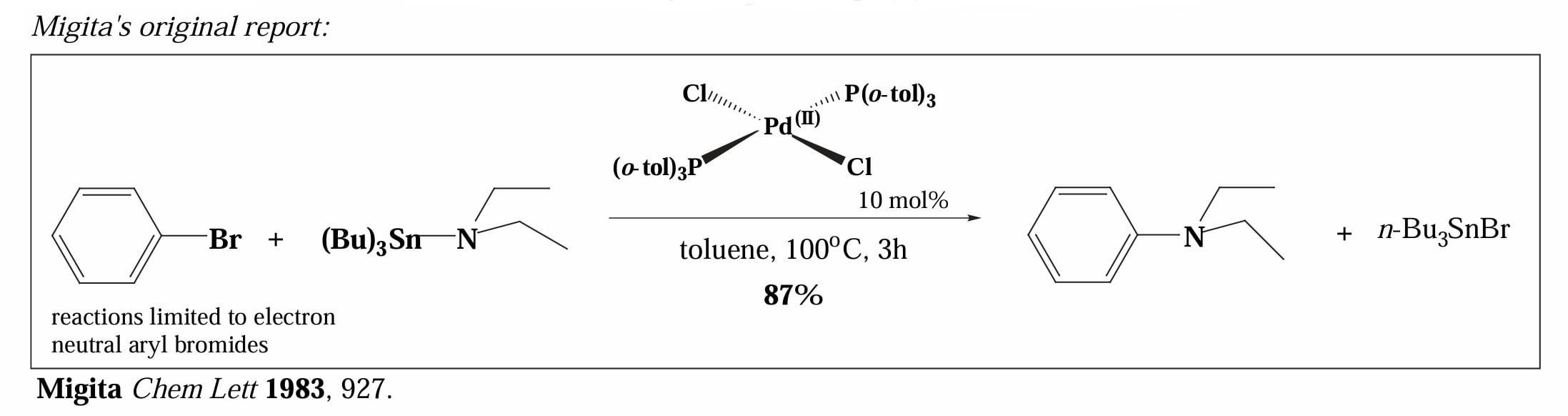
但是他的反应存在显著问题,仅有电中性的芳基溴底物能够得到很好的收率,且胺基锡试剂不稳定,这些都限制了反应的实际应用。
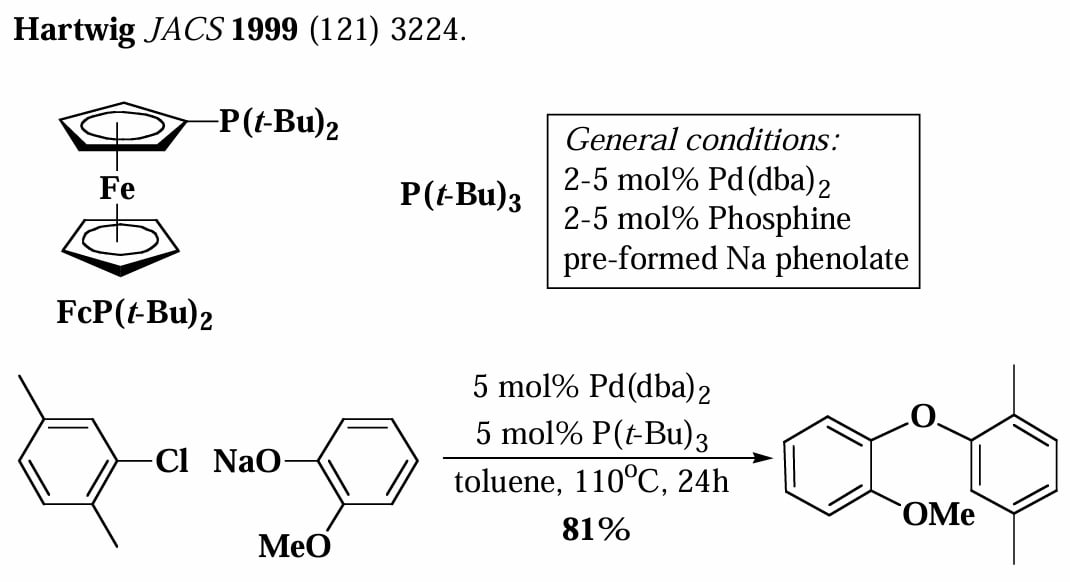
1995年,Buchwald 和 Hartwig 分别独立发现在大位阻碱存在下,芳基溴和游离胺可以直接发生偶联反应,无需使用有机锡试剂。
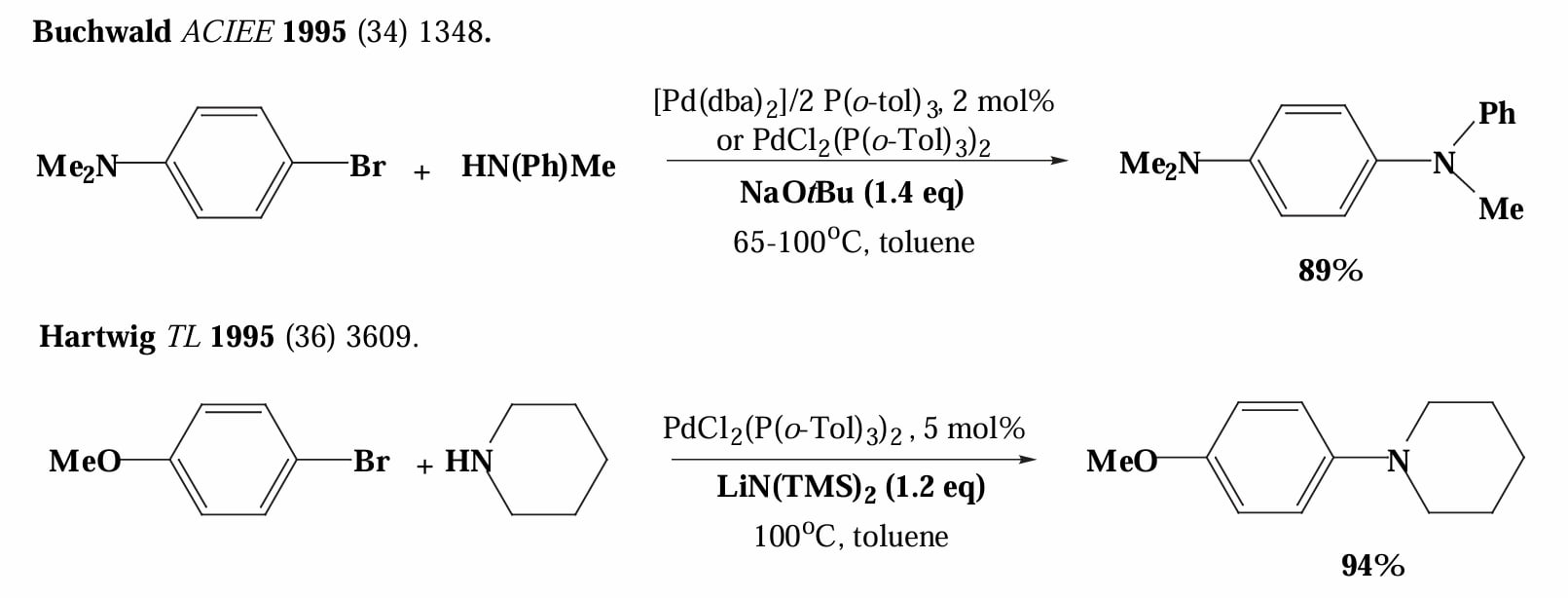
大位阻碱的作用是拔去游离胺的氢,同时脱去卤素。
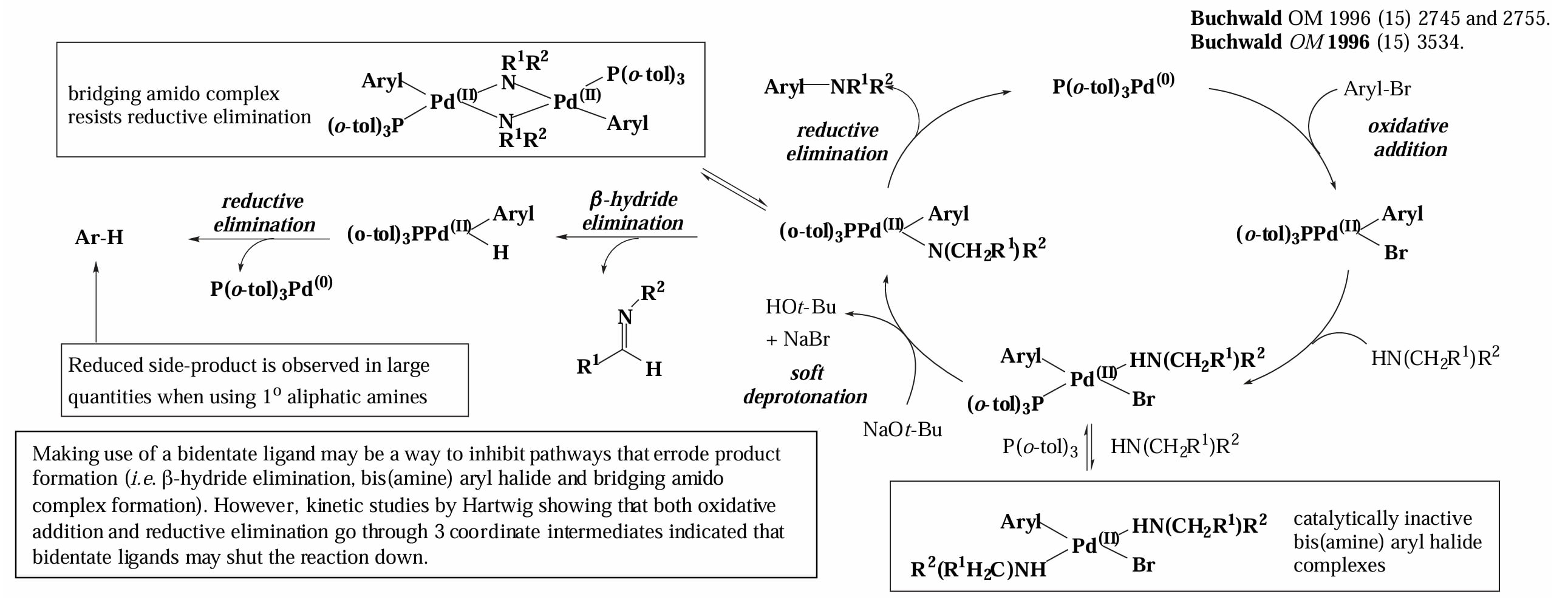
如果胺具有 α-H,那么可能存在 β-H elimination 的副反应,生成亚胺后再发生还原消除得到芳烃。
反应对于不同的配体表现出细微的差异,对于单齿膦配体来说,反应经历一个三配位 Pd complex 中间体,这就导致其可能发生 β-H elimination 或者是催化剂的二聚,降低反应速率。
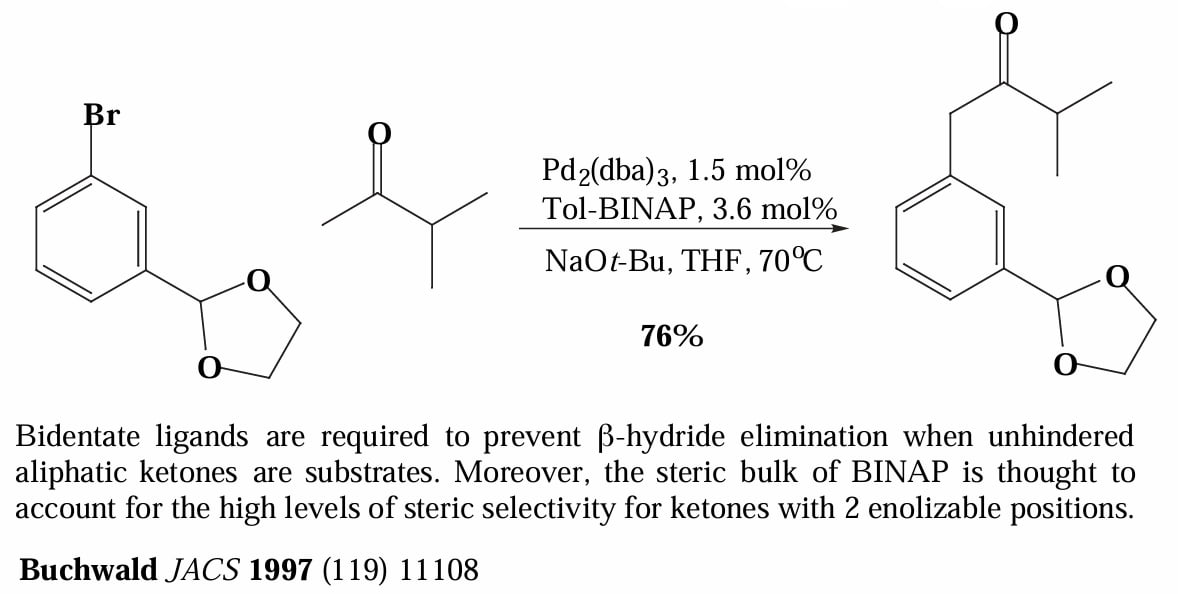
而对于双齿膦配体,反应经历一个四配位的 Pd complex 中间体,尽管发生 reductive elimination 的速率比三配位中间体稍慢,但是双齿配体占据了 Pd 的配位空位,因此极大的减少了 β-H elimination 和催化剂二聚的副反应。
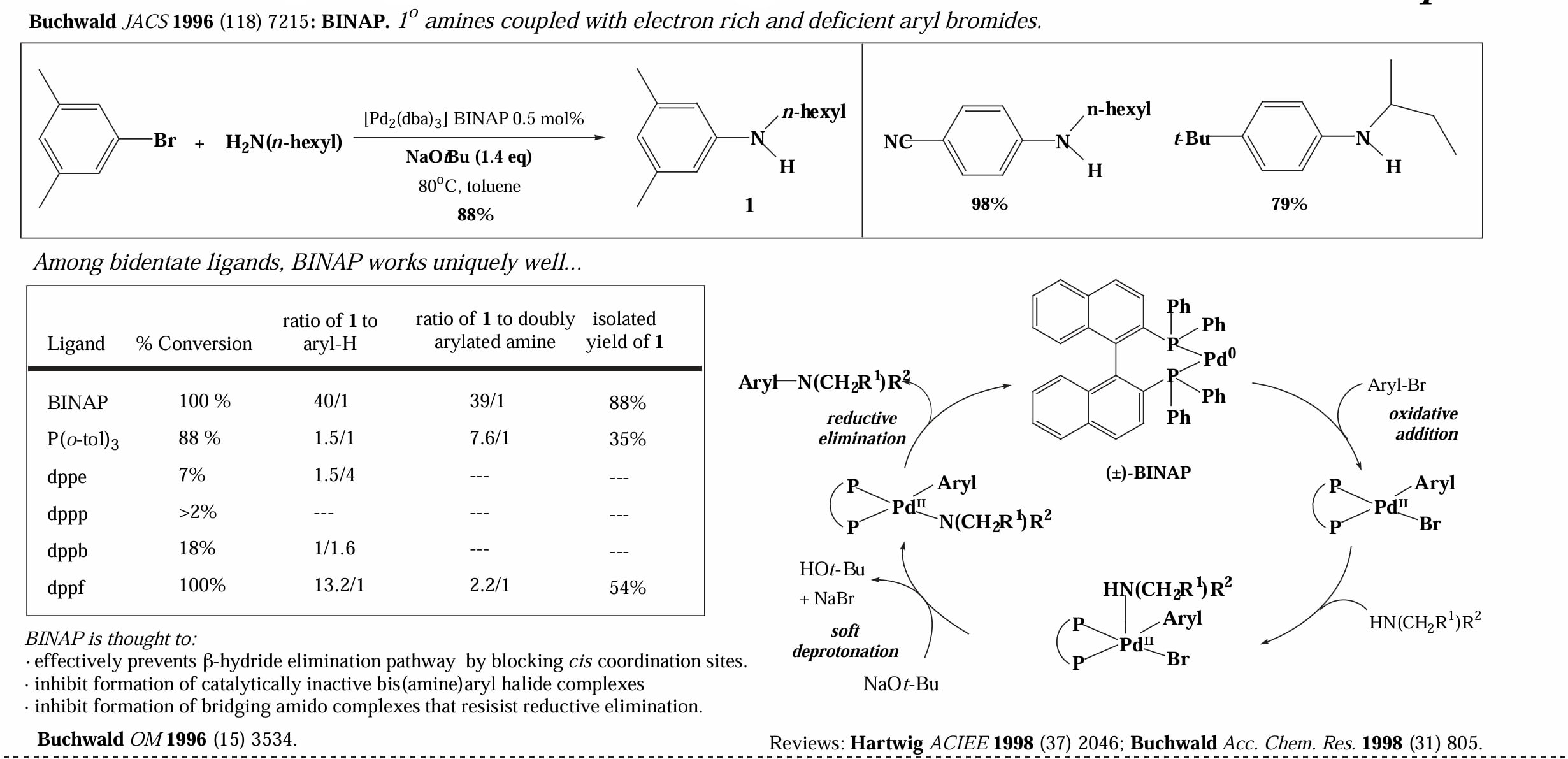
类似的,把亲核物种从胺换成烯醇负离子或者酚氧负离子可以实现 C-C/C-O 偶联。
3.9 Heck Reaction
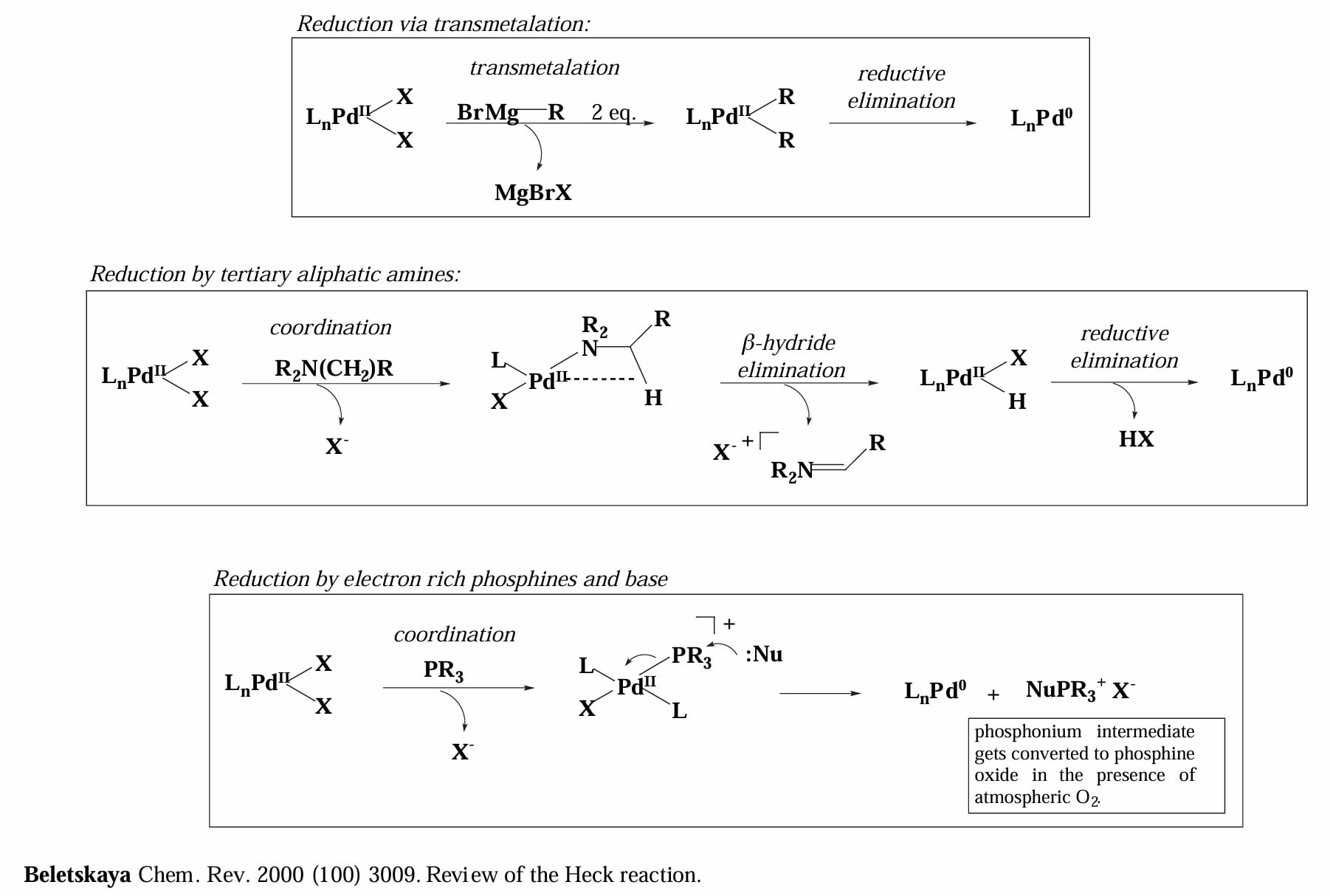
在进入反应之前先快速介绍一下从 Pd(II) 生成 Pd(0) 催化物种的几种路径。
首先是通过转金属+还原消除生成 Pd(0),例如常见的催化剂 会通过两次转金属生成 ,随后发生还原消除生成 Pd(0) 以及 R-R 烷烃。
其次是通过三级脂肪胺的 β-H elimination,常见例子如三乙胺;最后是利用富电子的膦配体或碱还原金属。
1982年,Heck 等人发现可以将烯基/芳基卤化物和烯烃直接连接,无需转金属试剂。反应选择性的得到 E 型烯烃。
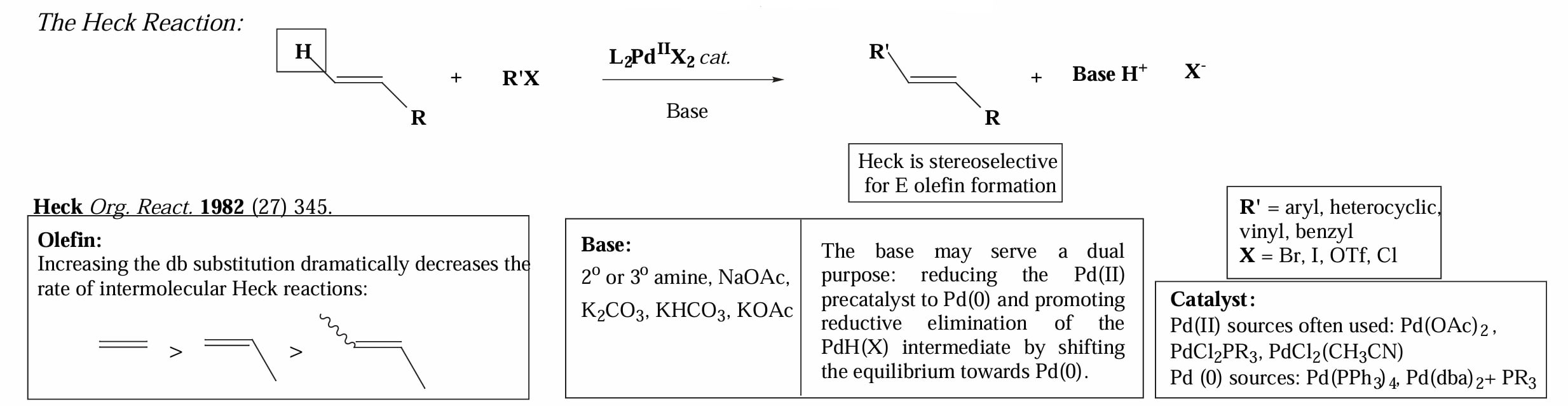
对于烯烃底物,位阻增大会显著影响反应速率;反应的碱的作用包括在一开始将 Pd(II) 转化为 Pd(0),同时将还原消除后的 物种转化为 Pd(0)。
Heck 反应的反应机理可以分为两种情况,其基本模式相同,不同之处是烯烃配位的 Pd complex 具有不同的电性。
当底物为烯基(芳基)氯/溴/碘时,由于 为 strong σ-donor,Pd-X 键不易断裂,因此催化剂会解离中性配体 L 后与烯烃配位,这种情况称为 neutral mechanism。
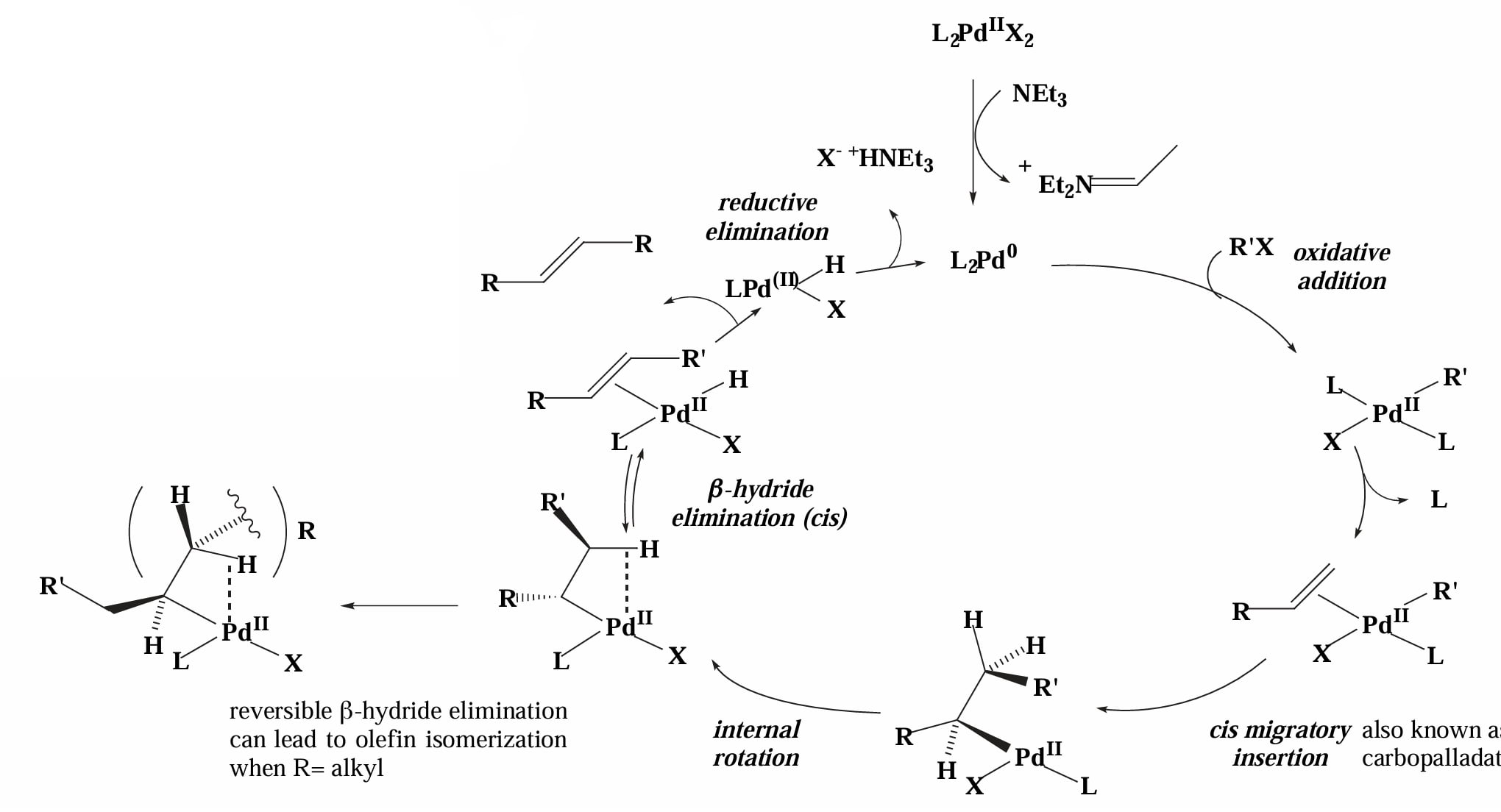
当 X=OTf, OAc 或存在 Ag/Tl 盐时,此时 Pd-X 将会断裂,催化剂解离负离子 后与烯烃配位,此时的 Pd complex 带正电,因此称为 cationic mechanism。
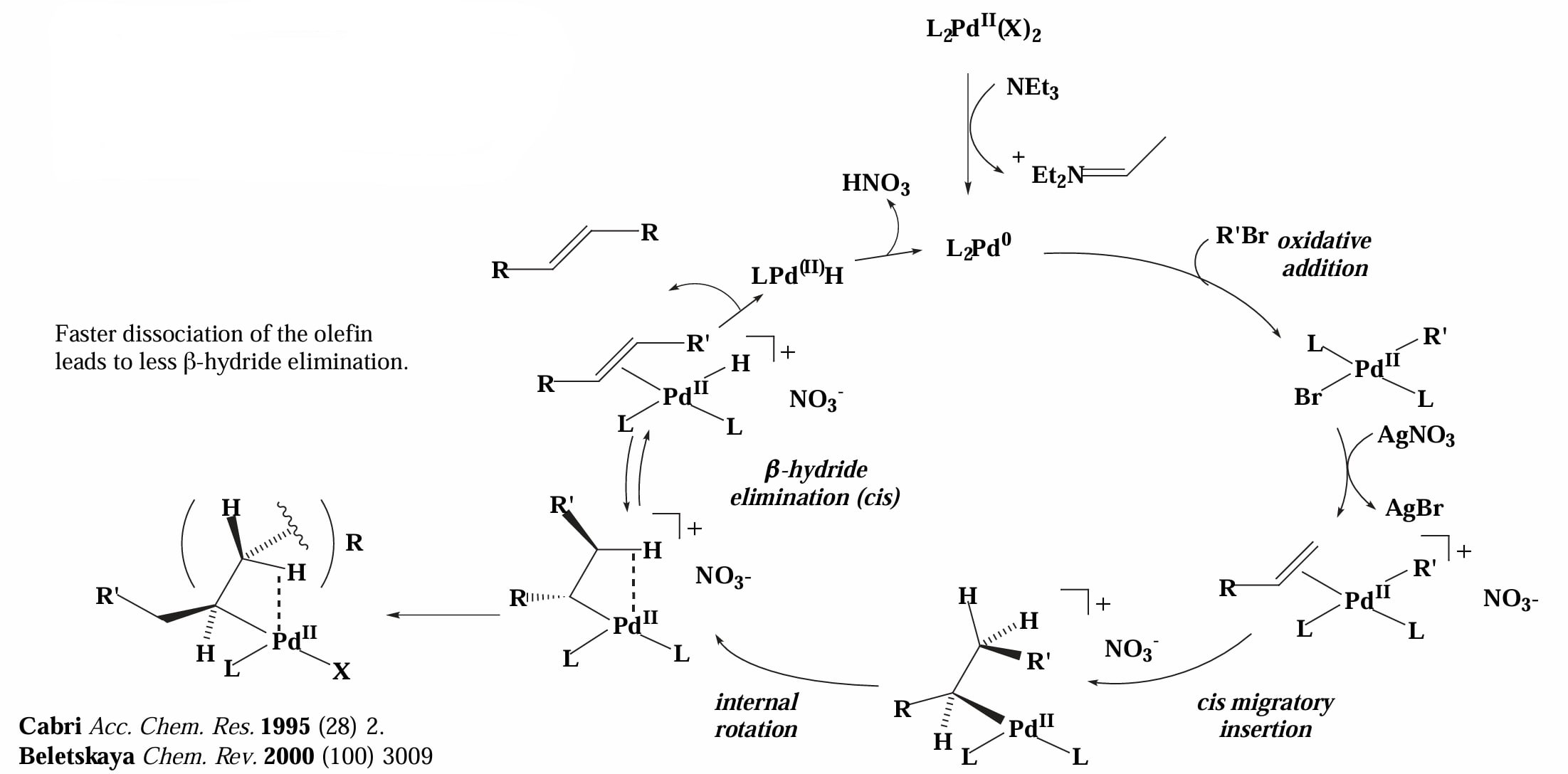
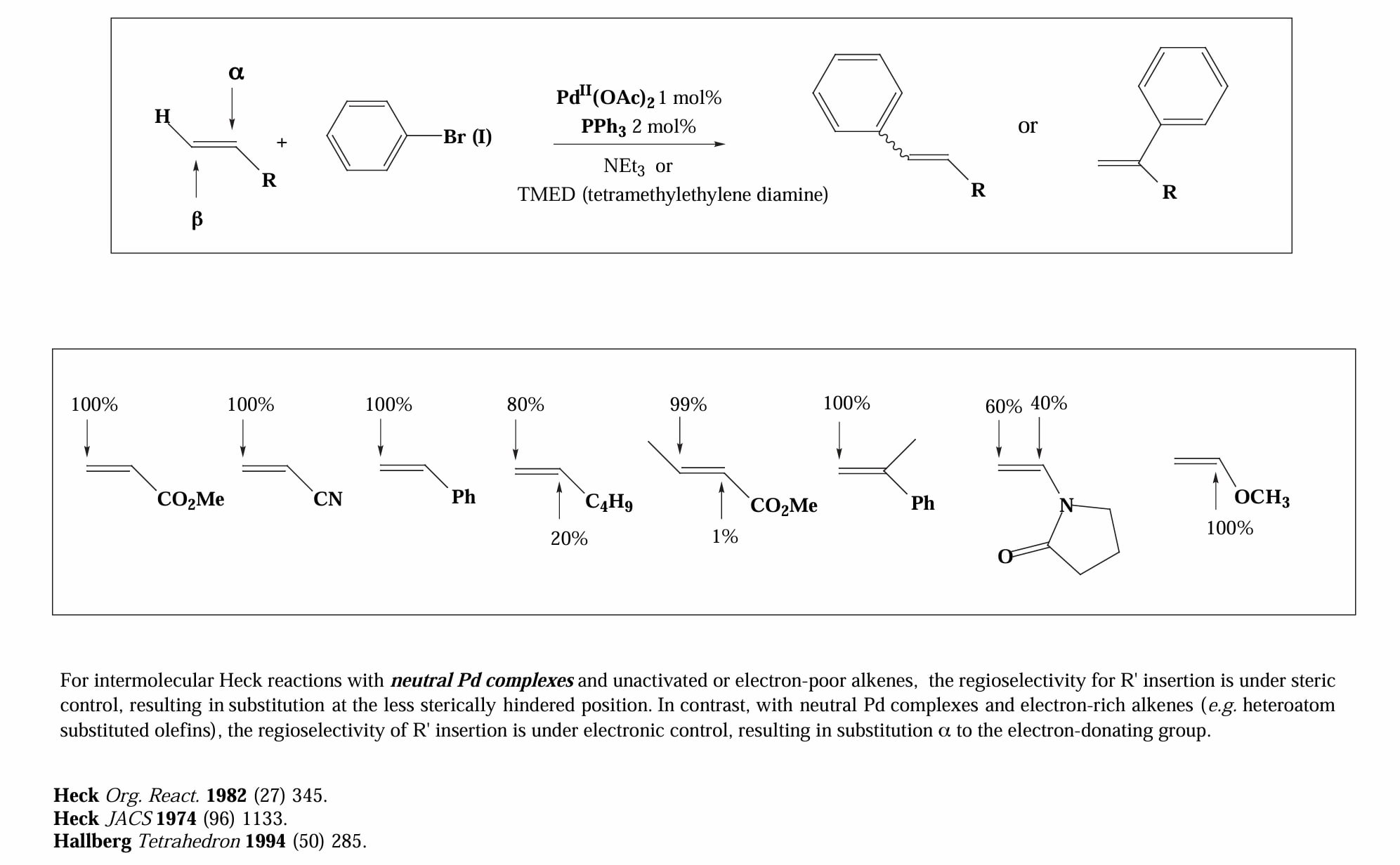
这两种不同的反应机理差异体现在反应的区域选择性上。对于中性机理,缺电子烯烃和中性烯烃的区域选择性由位阻控制,富电子烯烃的区域选择性由电性控制。
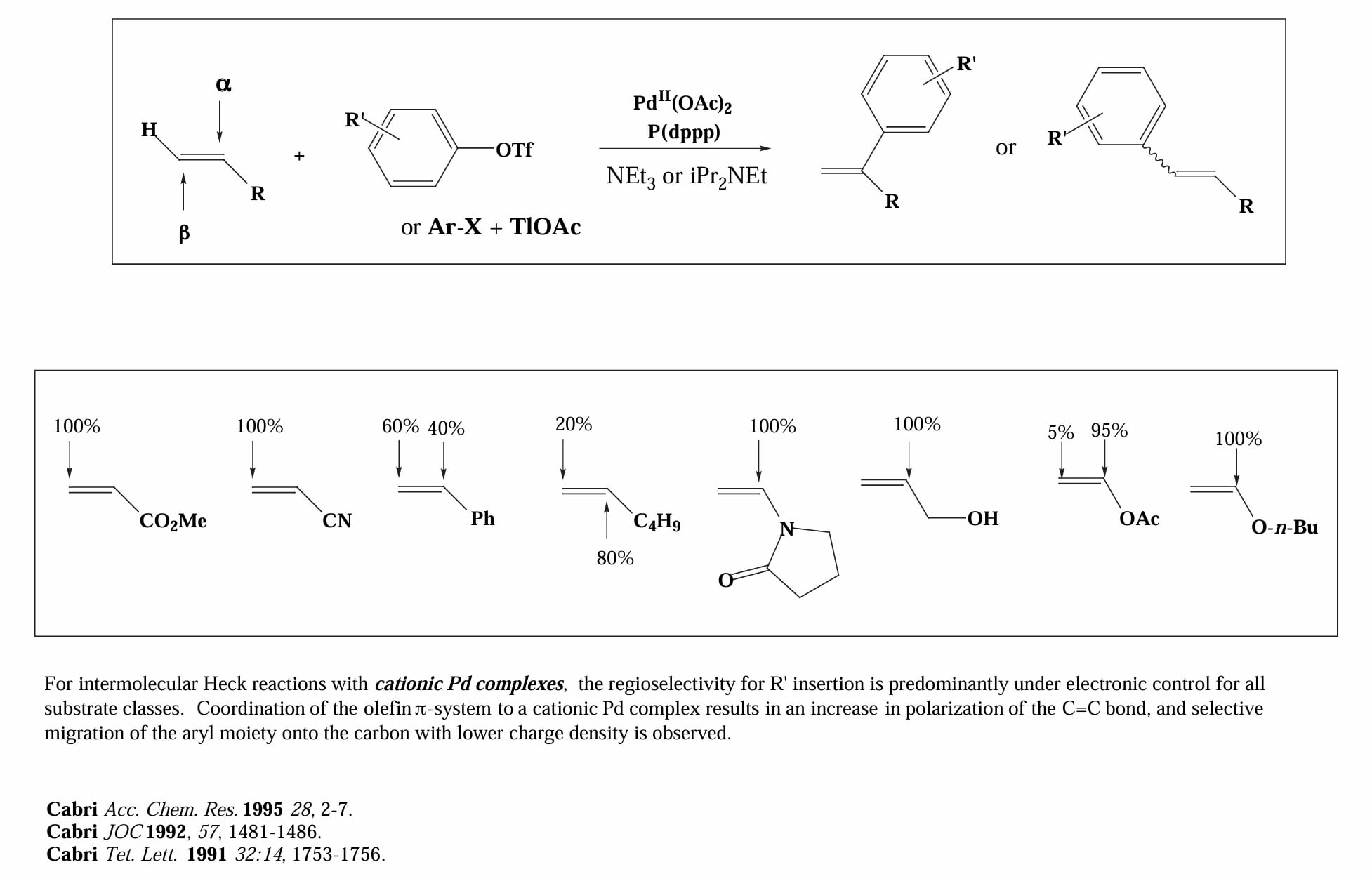
对于阳离子机理,几乎所有情况都由电性控制。
对于分子内反应还需要考虑环构象的影响,反应倾向于发生 exo-trig 形成5,6,7元的小环;而对于大环化合物则从位阻更小的一侧进攻,发生 endo-trig 反应。
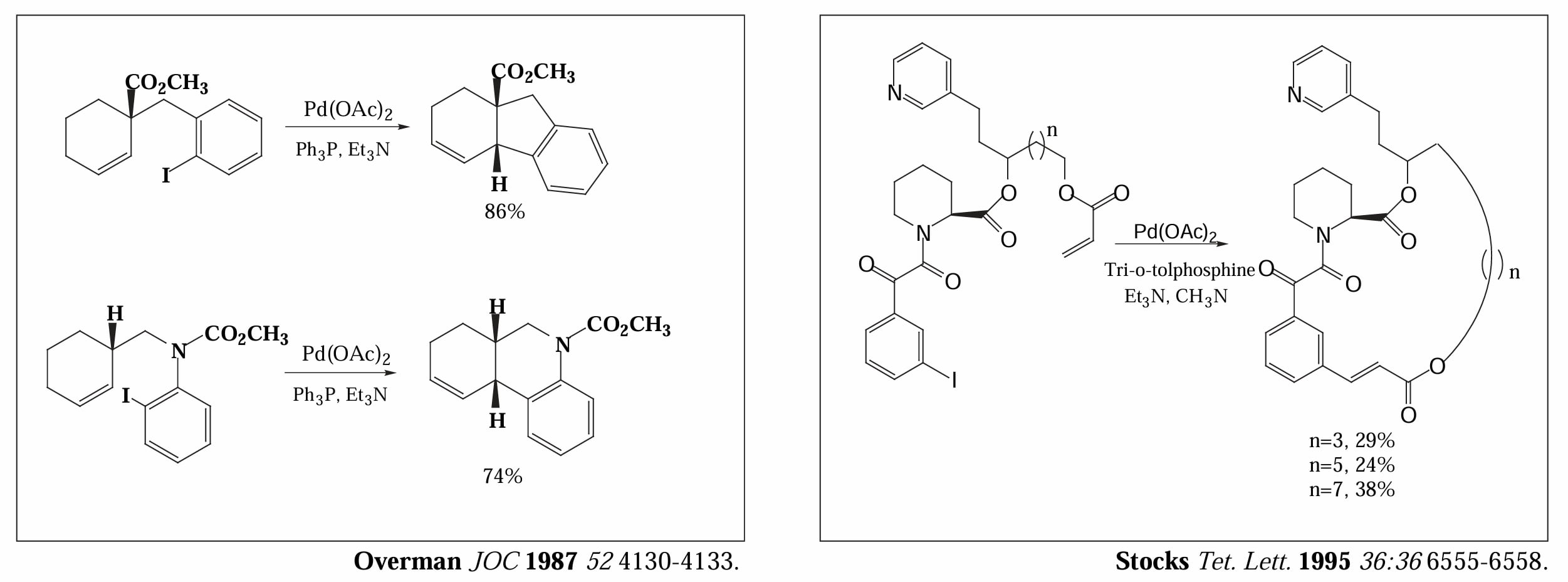
最后是两个例子:
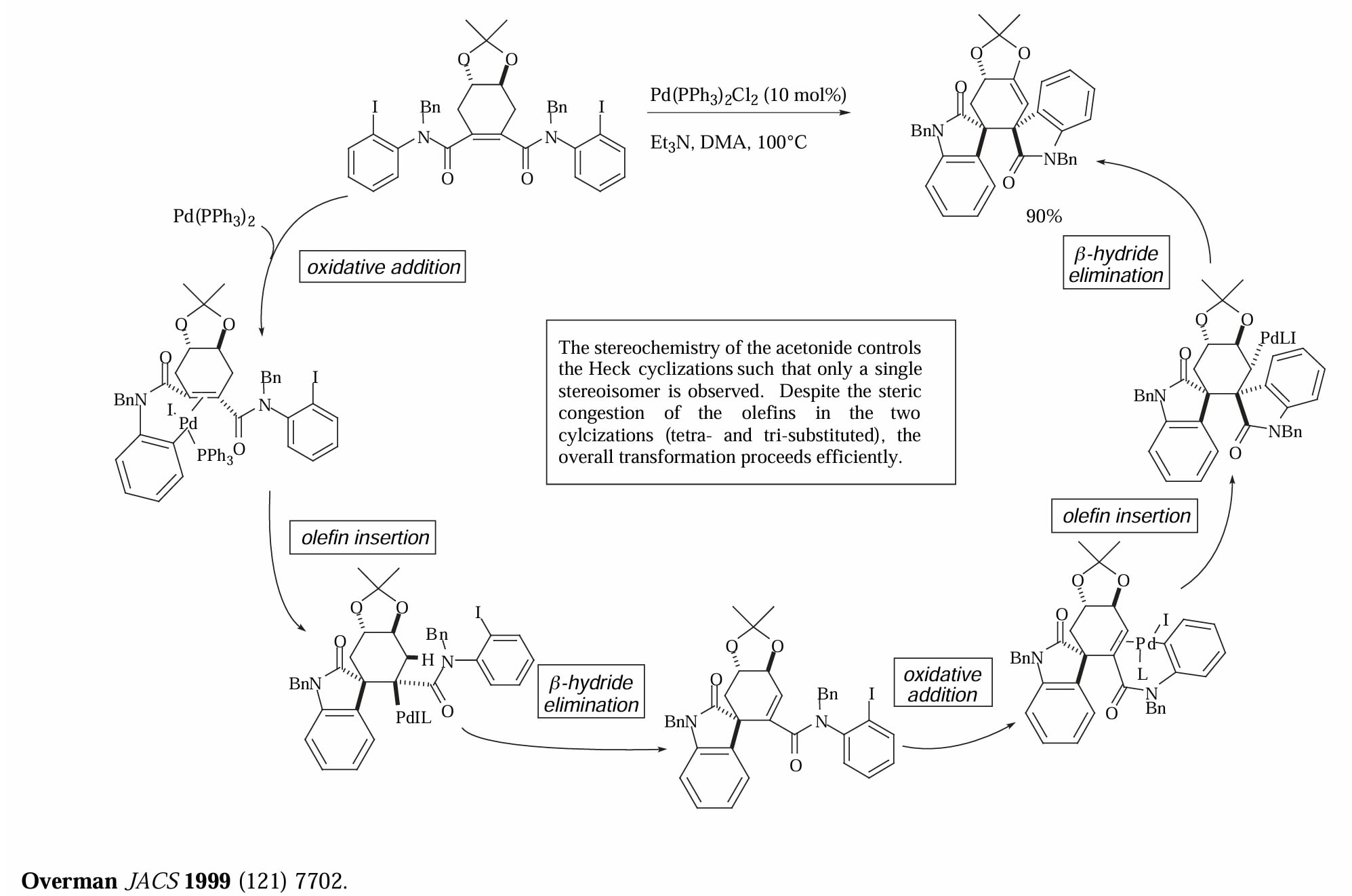
Overman 使用连续的 Heck 反应构建季碳中心。
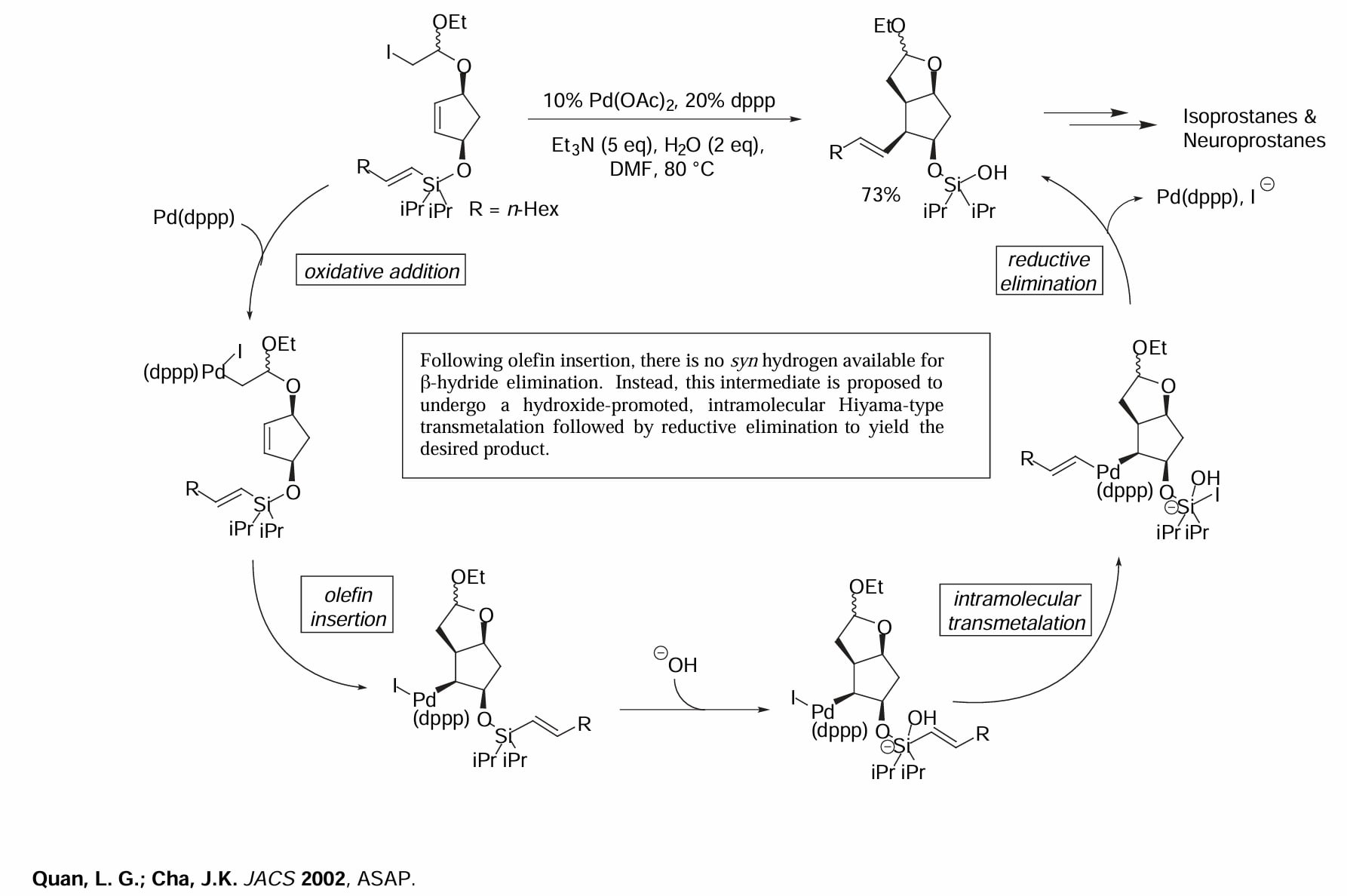
Quan 使用串联的 Heck-Hiyama 反应在双键两侧引入两个基团。
3.10 Summary
本节介绍了几个最为常见的人名反应,其在有机合成中几乎随处可见,这些反应为复杂分子的合成提供了强有力的工具。
3.11 Reference
- 1.Uchino, M.; Asagi, K.; Yamamoto, A.; Ikeda, S. Preparation and Properties of Aryl(Dipyridyl)Nickel Halide Complexes. Journal of Organometallic Chemistry 1975, 84 (1), 93–103. ↩
- 2.Yoshikai, N.; Mashima, H.; Nakamura, E. Nickel-Catalyzed Cross-Coupling Reaction of Aryl Fluorides and Chlorides with Grignard Reagents under Nickel/Magnesium Bimetallic Cooperation. J. Am. Chem. Soc. 2005, 127 (51), 17978–17979. ↩
- 3.Selig, R.; Schollmeyer, D.; Albrecht, W.; Laufer, S. The Application of Stille Cross-Coupling Reactions with Multiple Nitrogen Containing Heterocycles. Tetrahedron 2011, 67 (47), 9204–9213. ↩
- 4.(1) Casado, A. L.; Espinet, P. Mechanism of the Stille Reaction. 1. The Transmetalation Step. Coupling of R1 I and R2 SnBu3 Catalyzed by Trans -[PdR1 IL2 ] (R1 = C6 Cl2 F3 ; R2 = Vinyl, 4-Methoxyphenyl; L = AsPh3 ). J. Am. Chem. Soc. 1998, 120 (35), 8978–8985. (2) Casado, A. L.; Espinet, P.; Gallego, A. M. Mechanism of the Stille Reaction. 2. Couplings of Aryl Triflates with Vinyltributyltin. Observation of Intermediates. A More Comprehensive Scheme. J. Am. Chem. Soc. 2000, 122 (48), 11771–11782. ↩
- 5.Farina, V.; Kapadia, S.; Krishnan, B.; Wang, C.; Liebeskind, L. S. On the Nature of the “Copper Effect” in the Stille Cross-Coupling. J. Org. Chem. 1994, 59 (20), 5905–5911. ↩
- 6.Han, X.; Stoltz, B. M.; Corey, E. J. Cuprous Chloride Accelerated Stille Reactions. A General and Effective Coupling System for Sterically Congested Substrates and for Enantioselective Synthesis. J. Am. Chem. Soc. 1999, 121 (33), 7600–7605. ↩
- 7.Harris, D. H.; Lappert, M. F. Monomeric, Volatile Bivalent Amides of Group IVB Elements, M(NR12)2 and M(NR1R2)2(MGe, Sn, or Pb; R1Me3Si, R2Me3C). J. Chem. Soc., Chem. Commun. 1974, No. 21, 895–896. ↩
- 8.Kira, M.; Sato, K.; Sakurai, H.; Hada, M.; Et., A. Ab Initio MO Study of the Reaction of Pentacoordinate Allylsilicates with Aldehydes. Chemistry Letters 1991. ↩
- 9.Miyaura, N.; Yamada, K.; Suzuki, A. A New Stereospecific Cross-Coupling by the Palladium-Catalyzed Reaction of 1-Alkenylboranes with 1-Alkenyl or 1-Alkynyl Halides. Tetrahedron Letters 1979, 20 (36), 3437–3440. ↩
- 10.Uenishi, J.; Beau, J. M.; Armstrong, R. W.; Kishi, Y. Dramatic Rate Enhancement of Suzuki Diene Synthesis. Its Application to Palytoxin Synthesis. J. Am. Chem. Soc. 1987, 109 (15), 4756–4758. ↩
- 11.Synthesis of 2-Dienylindole, SB 242784, by a Three-Component Palladium-Catalyzed Coupling Reaction. Tetrahedron Letters 1998, 39 (51), 9347–9350. ↩