《ART》章末总结6
《ART》第六章,本章是金属有机反应,不过本书内容着重在反应模式和机理书写上,更多细节内容需要参考更高级的书目。
6.1 Introduction to the Chemistry of Transition Metals
Many widely used organic reactions require the use of transition metals. This chapter discusses some of the typical reactions of transition metals. These principles will then be applied to understanding some of the organic transformations mediated by these metals.
( 本书主要目的是为了让读者能够合理的写出陌生反应的机理,因此在章节编排时不是按照反应机理类别或是金属类别分类,而是根据总体反应类型分类。同时金属有机化学是一个非常广泛的话题,本书无法做到面面俱到,因此部分金属有机反应没有提及或是一笔带过,同时立体化学不是该书重点,因此更深入的讨论需要参考其他金属有机教材。 )
6.1.1 Conventions of Drawing Structures
The conventions for drawing organometallic compounds differ in subtle ways from those used to to draw “ordinary” organic compounds. The most important difference is the way in which bonds are drawn. In organic compouds, one does not use a line to connect a bond to an atom. In organmetallic and inorganic compounds, however, a line sometimes connects an atom and a or bond. In this case, the line indicates that the pair of electrons in the or bond is shared with the metal also.
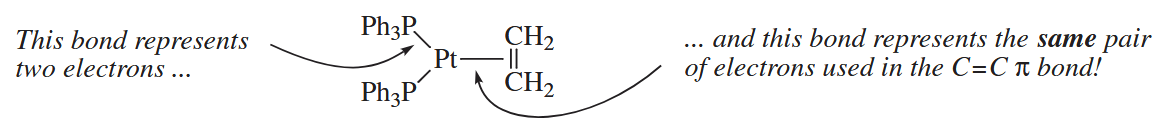
An even more confusing situation arises in complexes in which the electrons in a system spread over three or more atoms are used to make a bond to a metal.
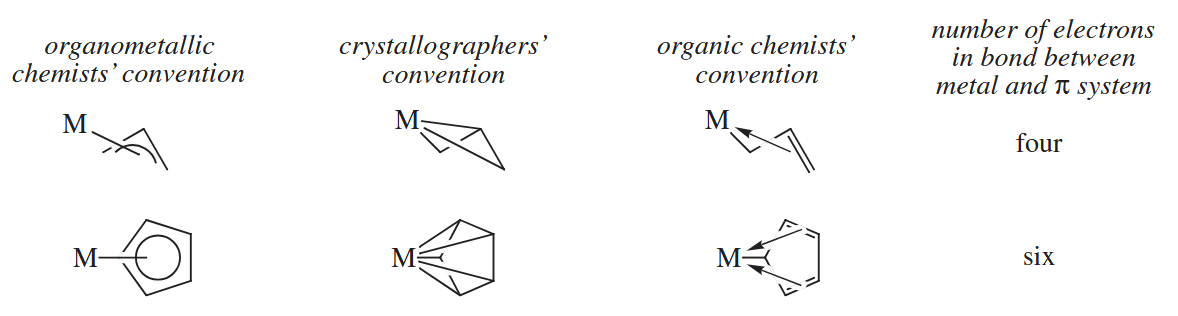
( 其实上图中第一种表示不论在什么领域中都相当常见,反而是所谓的 organic chemists’ convention 几乎没有人用。 )
Formal charges are usually omitted in inorganic and organometallic complexes. The acid-base bond is sometimes indicated by an arrow pointing from the ligand to the metal, but more often it is indicated by an ordinary line.
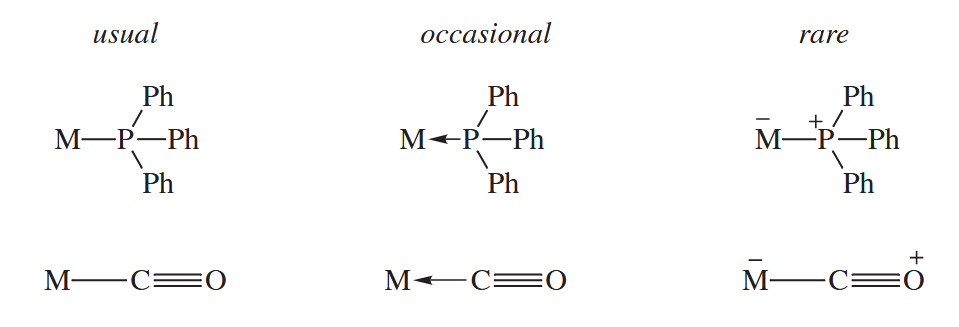
6.1.2 Counting Electrons
The chemistry of any particular metal complex can be understood by examing its total electron count, its number of d electrons, and the metal’s oxidation state.
( 数电子非常基础但又非常关键,不过限于篇幅,本节只做简单叙述。 )
Transition metals have 10 valence AOs, one s, five d, and three p, in that order, and they follow the 18-electron rule. The 18-electron rule is much less rigorous for transition metals than the octet rule is for main-group elements.
Ligands attached to metals can provide anywhere from one to six electrons to the metal.
- Monovalent groups such as alkyl, alkoxy, H, , halogen, etc.
- Divalent fragments such as carbenes or alkylidene groups, imido groups, or oxide.
- Lewis bases such as are also 2-electron donors.
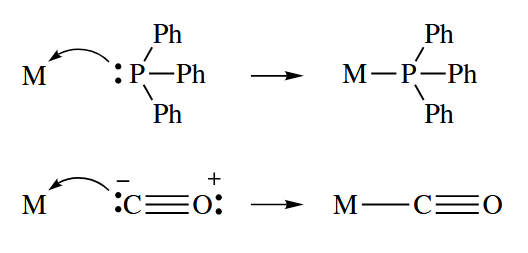
- Alkenes and alkynes can also act as Lewis bases toward metals by using the two electrons in their π bonds. Even σ bonds such as H-H and C-H bonds can act as two-electron donors to metals.
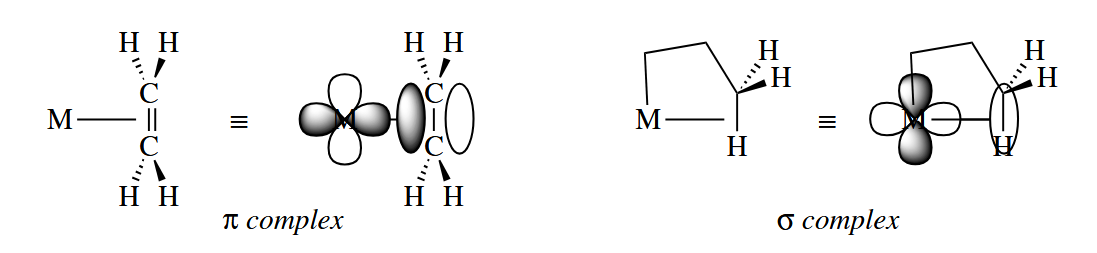
- Trivalent fragments or the allyl group can be a three-electron donor.
- Dienes are four-electron donors. The cyclopentadienyl group () is a five-electron donor. Benzene rings can act as six-electron donors.
Alkenes and alkynes have special characteristics as Lewis base donors. The face-on interaction of the π bond of an alkene with a p or d orbital on the metal produces a σ bond. And if the metal has a pair of electrons in another d orbital, the d orbital can overlap with the empty π* orbital of the alkene,
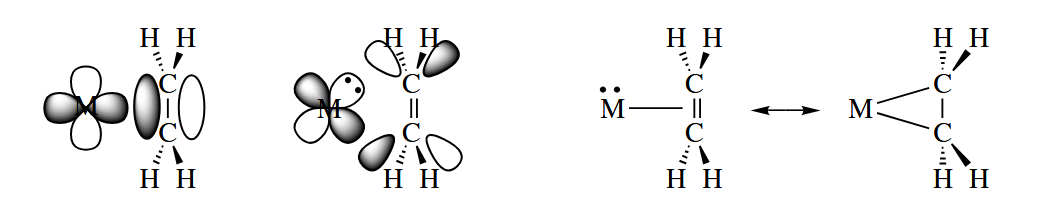
( 具有 π 键的化合物不仅可以用 π 电子填入金属的空轨道,同时还能用 反键轨道接受金属的一对孤对电子,其轨道对称性可以用 Dewar-Chatt-Duncanson 模型来解释。这种反键作用会削弱 C-C 键,但是也会增加 M-C 键强度。除了烯烃以外,CO 配体是另一个很好的例子。 )
A similar picture can be drawn for σ complexes.
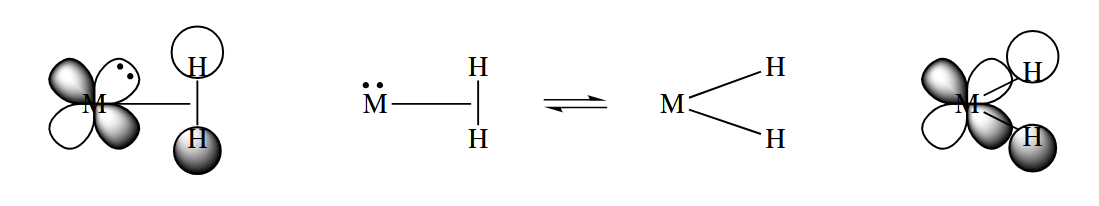
A scecond important determinant of the reactivity of a metal is its oxidation state. The assumption in calculating a metal’s oxidation state is that groups attached to the metal are more electronegative than the metal, and so all σ bonds are ionic bonds. If both electrons in every σ bond “belong” to the ligand, the metal is left with a charge, and that is its oxidation state.
In practice, you can determine the oxidation state by counting the number of covalent bonds to the metal.
The d electron count of the metal is calculated by substracting the metal’s oxidation state from the number of valence electrons (including the two s electrons).
( 注意这里的 d 电子数实际上包括了 s 电子,应当理解为价电子数更好。举个例子,对于 Fe(II) 来说,其 d 电子数为6,似乎没什么问题,但是对于 Fe(0) 来说,其 d 电子数为8,因为两个 s 电子也一并算在内。 )
6.1.3 Typical Reactions
Almost all of the reactions of metals can be classified into just a few typical reactions, and the reactions that metals promote in organic chemistry are simple combinations of these typical reactions.
The typical reactions of metal complexes are ligand addition/ligand dissociation/ligand substitution, oxidative addition/reductive elimination, insertion/β-elimination, α-insertion/α-elimination, σ-bond metathesis (including transmetallations and abstraction reactions), [ 2 + 2 ] cycloaddition, and electron transfer.
- Ligand addition and dissociation reactions are simple Lewis acid-base reactions. Many metal catalysts undergo a dissociation or association reaction to form the actual active species.
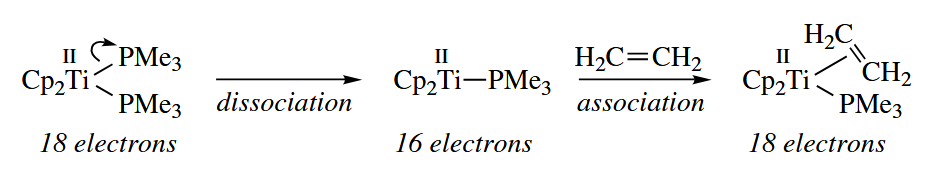
The deprotonation of M-H to give and is one quite common “dissociation” reaction in which the pair of electrons of the bond goes to the metal.
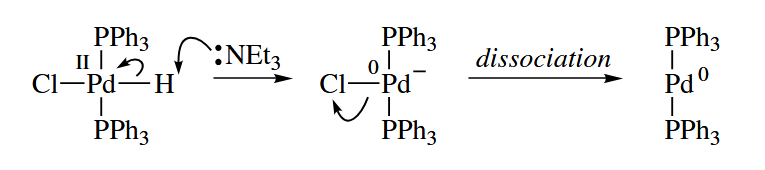
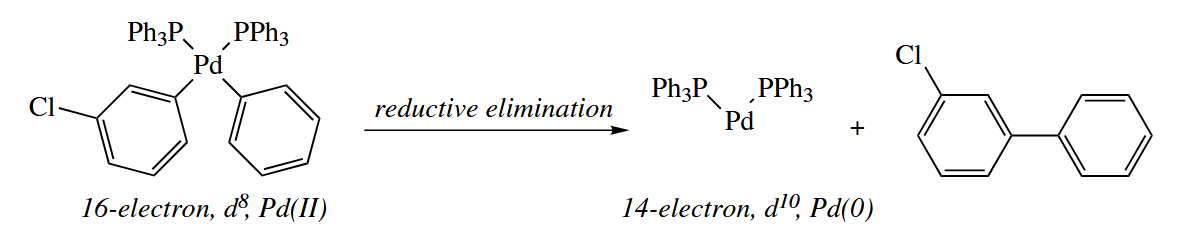
- Oxidative addition and reductive elimination are the microscopic reverse of each other.
In oxidative addition, a metal inserts itself into an X-Y bond. The reaction is an oxidation, because the metal’s oxidation state increases by 2, but the metal also increases its total electron count by 2, so it because less electron-deficient.
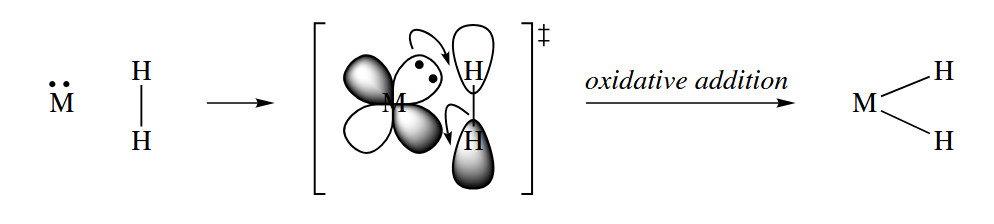
Oxidative additions can occur at all sorts of X-Y bonds, but they are most commonly seen at H-H (also H-Si, H-Sn, Si-Si, B-B, or other electropositive elements) and carbon-halogen bonds. Oxidative addition is very common for late metals like Pd, Pt, Ir, Rh, and the like.
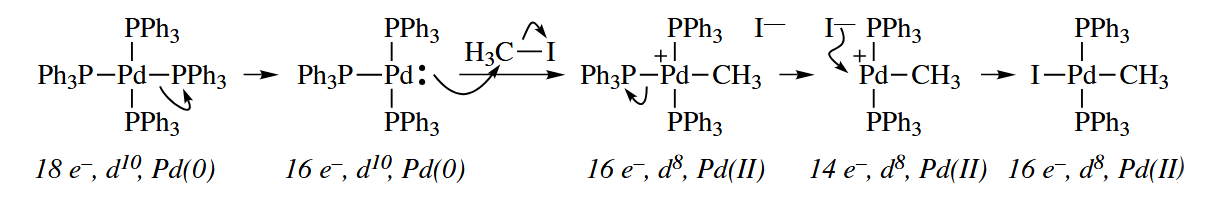
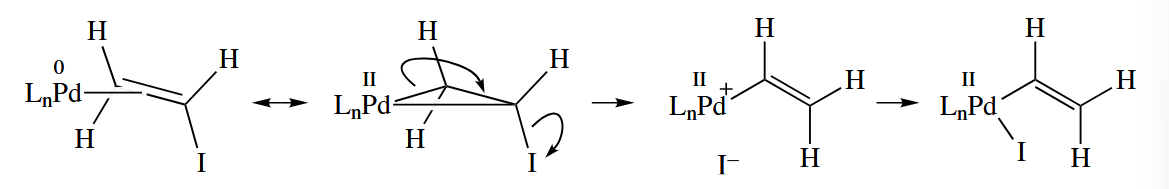
Oxidative addition can also occur at C()-X bonds. The mechanism is possiblity. Another possibility is that Pd coordinates to form a π complex. An electrocylic ring opening of the metallacyclopropane can then occur, and leaves to give a new cationic Pd-vinyl complex. When associates with the Pd, the overall result is oxidative addition.
- Reductive elimination is the microscopic reverse of oxidative addition. This reaction is usually most facile when the X-Y bond is strong. It’s known that the two groups must be adjacent to each other in the metal’s coordination sphere.
- Insertions and β-eliminations are also the microscopic reverse of each other. In an insertion, an A=B π bond inserts into an M-X bond. Insertion is usually proceeded by coordination of the A=B π bond to the metal, so it is sometimes called migratory insertion.
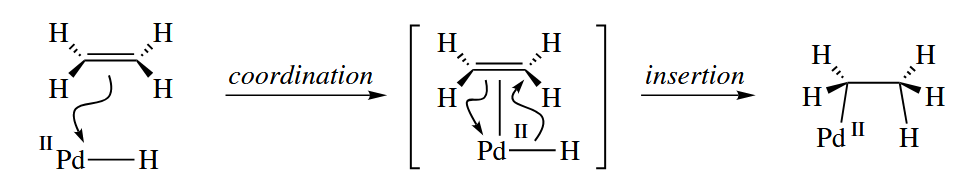
β-Elimination is the microscopic reverse of insertion. By far the most common β-elimination is the β-hydride elimination.
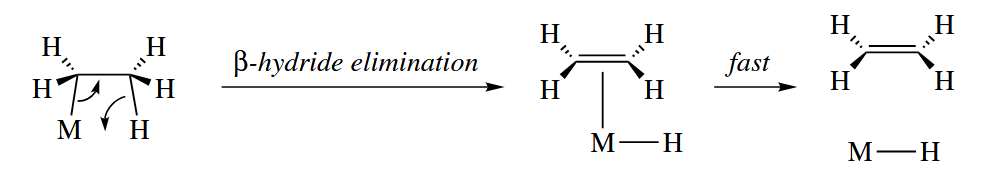
Insertions are generally thermodynamically favorable, providing a new σ bond at the expense of a π bond. β-Eliminations are very fast reactions, however, so insertions are readily reversible even when they are thermodynamically favorable.
Carbenoid species such as CO and RNC also undergo insertions and elimination. There are 1,1-insertions, as opposed to the more common 1,2-insertions. Again, the insertion is reversible.
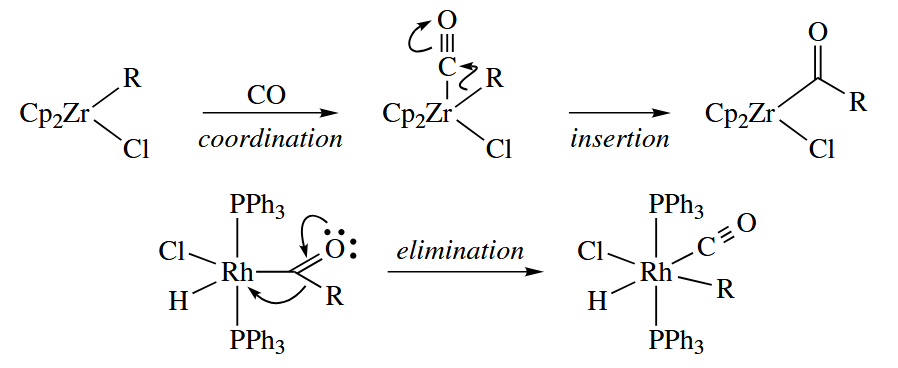
- α-Insertions and α-eliminations are considerably less common than migratory insertions and β-eliminations. In an α-insertion, a ligand on a metal migrates to an adjacent atom that is doubly bound to the metal, and the electrons of the π bond migrate to the metal.
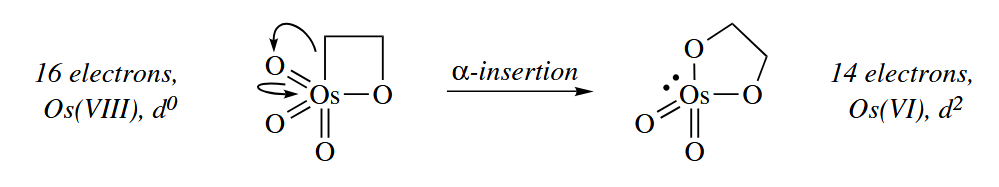
α-Insertions and α-eliminations are most commonly seen in third-row transition metals, which are more likely to form multiple bonds.
( α-消除和上文的羰基插入的区别在于:α-消除中底物为 M=Y 双键,且基团迁移把电子推向金属;羰基的插入则是以 M-CO 为底物,基团迁移把电子推向氧原子。这一点差异导致羰基的插入不会影响金属的电子数和氧化态(忽略 CO 配位的瞬态过程),而 α-消除会降低金属氧化态。 ) - Sigma-bond metathesis reactions involve the swapping of M-X and Y-Z σ bonds to give M-Z and X-Y (or M-Y and X-Z). The reaction is concerted, involving a four-center transition state.
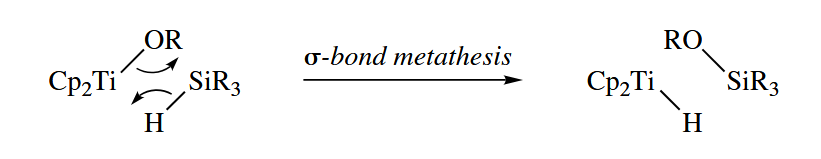
For this reaction is seen espcially often among early, transition metals. (In metals with a or greater electron count, a two-step oxidative addition-reductive elimination pathway can be invoked to explain the same overall reaction.)
Transmetallations, in which M-X and M’-Y swap partners to give M-Y and M’-X, are a special kind of σ-bond metathesis reaction.
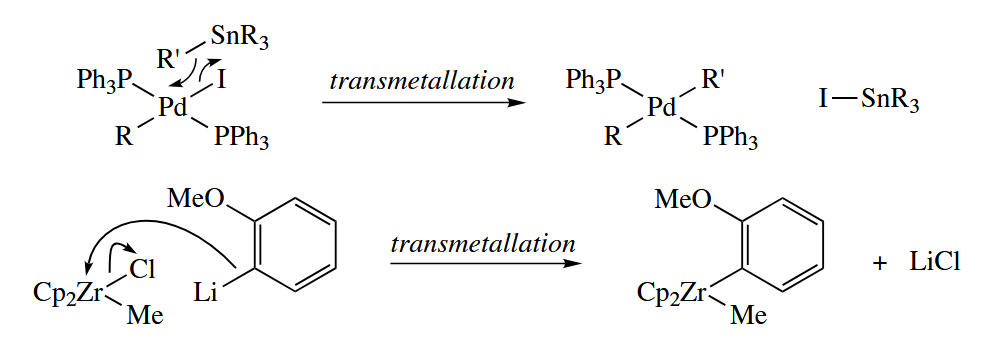
Abstraction reactions are four-center, concerted processes that are closely related to σ-bond metathesis. β-Abstraction occurs most often in metal dialkyl complexes such as .
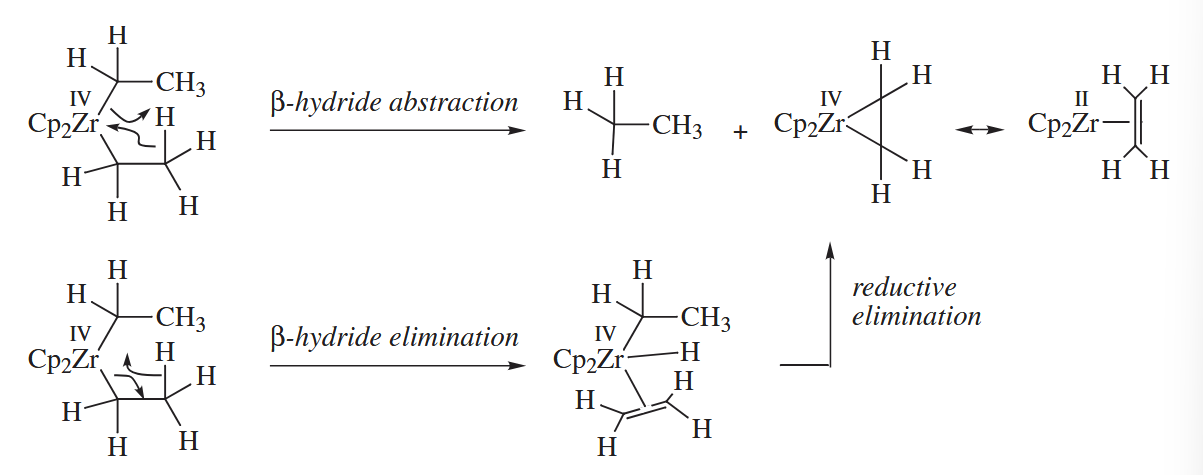
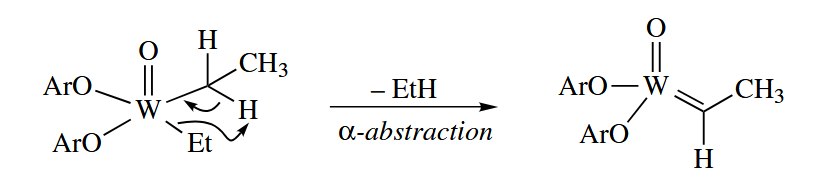
γ-Abstractions have also been observed, although they are much rarer. α-Abstraction reactions are especially common among third-row transition metals, which readily form multiple bonds to C.
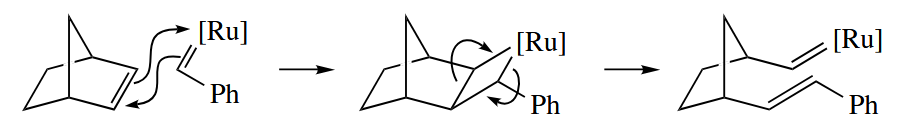
- The [ 2 + 2 ] cycloaddition is another typical reaction of metal (Chapter 4).
- Metals also undergo one-electron transfer reactions (Chapter 5).
6.1.4 Stoichiometric vs. Catalytic Mechanisms
Whether a reaction requires stoichiometric or catalytic quantities of a transition metal has a strong bearing on how one draws a mechanism for the reaction. The mechanism of a reaction that requires stoichiometric quantities of the metal can be written in a linear fashion like a polar or pericyclic mechanism. However, the mechanisms of metal-catalyzed reactions are usually drawn in a circular fashion, showing how the original metal species is regenerated at the end of each catalytic cycle.
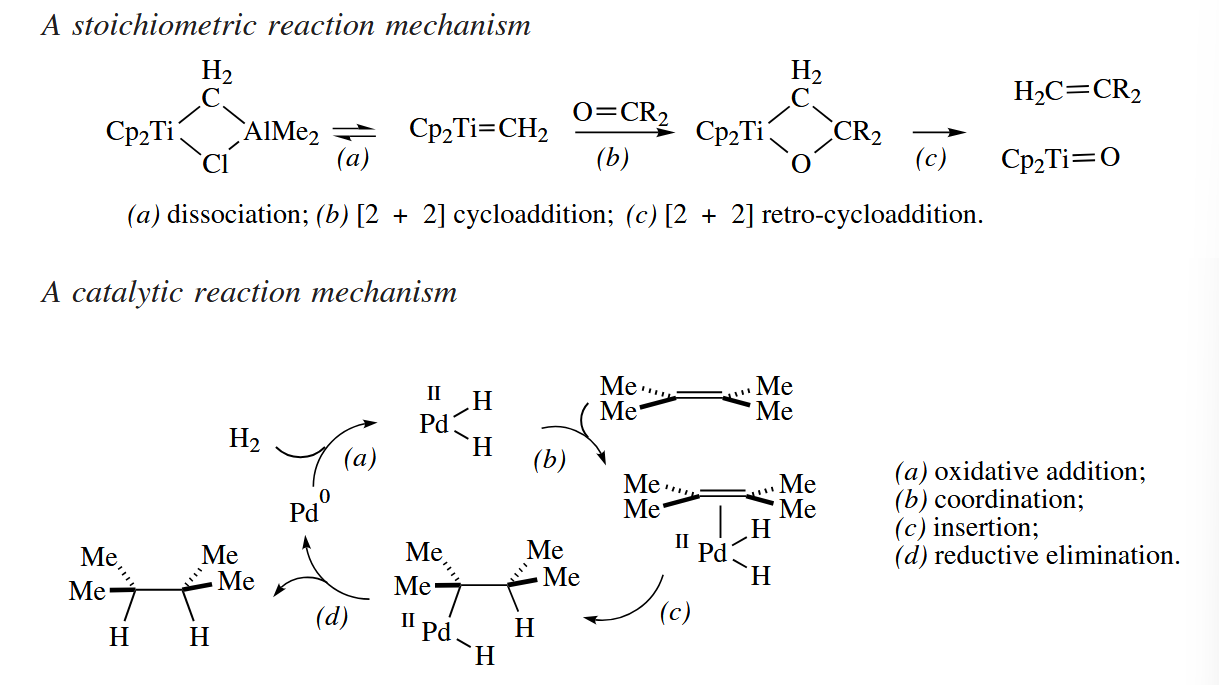
Because it is not the case that every line represents exactly two electrons in a drawing of an organometallic or inorganic compound, it follows that the curved-arrow convention for showing the movement of electrons cannot be applied unambiguously when reaction mechanisms involving transition metals are drawn.
You may use curved arrow to show electrons movement in some steps if you wish, but it is more important for you to name every step.
Organometallic and inorganic catalysts are often classified as homogeneous or heterogeneous. These two terms simply mean soluble or insoluble. Homogeneous catalysts are easier to study and often have more predictable behavior than heterogeneous catalysts. Heterogeneous catalysts tend to be used more in large-scale industrial synthesis.
6.2 Addition Reactions
6.2.1 Late-Metal-Catalyzed Hydrogenation and Hydrometallation (Pd, Pt, Rh)
One of the first metal-catalyzed reactions learned by organic chemistry students is the Pd-catalyzed hydrogenation of alkenes and alkynes. For a long time, the mechanisms of the heterogeneous hydrogenations were considered quite mysterious, but with the advent of homogeneous catalysts like Wilkinson’s catalyst, it became possible to study the mechanisms in detail.
Every late-metal hydrogenation catalyst, whether homogeneous or heterogeneous, probably uses exactly the same catalytic cycle, although some catalysts, particularly sterically encumbered ones such as Wilkinson’s catalyst, require that ligand dissociation or substitution occur before the catalytic cycle gets underway.
Formic acid and 1,4-cyclohexadiene are sometimes used as alternative sources in hydrogenation and hydrogenolysis reactions.
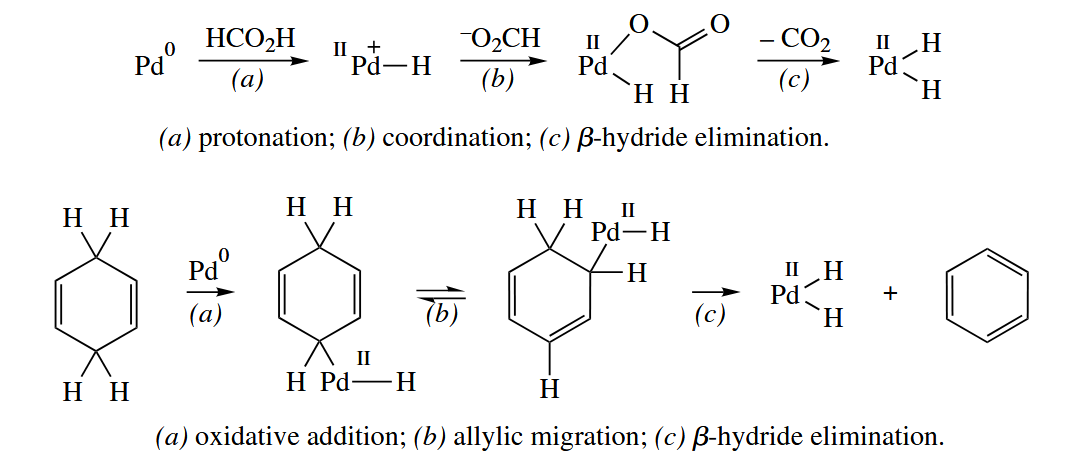
Late-metal complexes of Pd, Pt, and Rh can also catalyze hydrosilylation, hydrostannylation, hydroboration, and diborylation reactions of π bonds. The mechanisms of all these reactions are the same as hydrogenation.
After oxidative addition of an unsymmetrical σ bond (E-H) to the metal, an unsaturated compound can insert into either the M-E or the M-H bond. In any case, either pathway gives the same product after reductive elimination.
( 注意含 π 键化合物插入不同的键仅给出不同的反应机理,最终仍然会得到相同的化合物。 )
6.2.2 Hydroformylation (Co, Rh)
Rh and Co complexes catalyze hydroformylation, the addition of and CO to a C=C π bond to give an aldehyde. The hydroformylation of propene is used to make butyraldehyde, which is hydrogenated to give butanol, a widely used solvent.
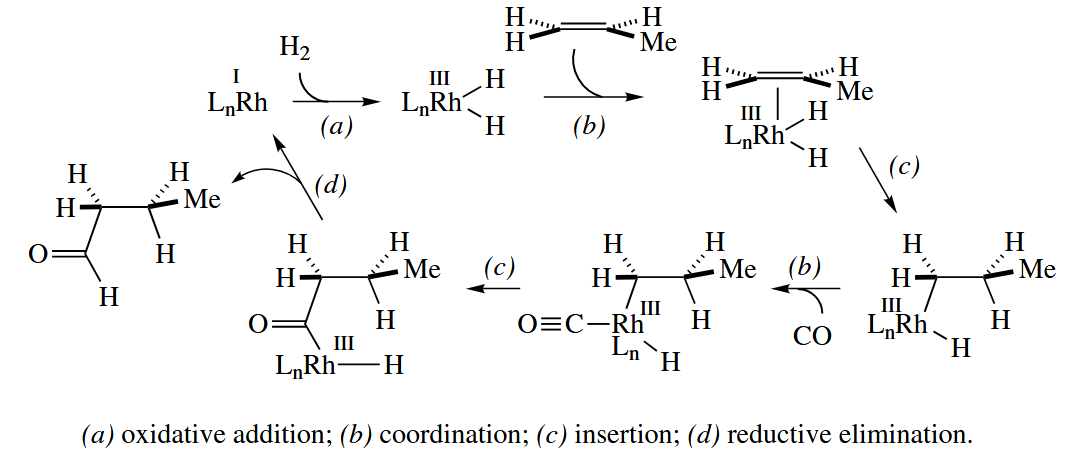
Insertion of propene into the M-H bond can give two isomers, one with a 1° alkyl–metal bond (as shown) and one with a 2° alkyl–metal bond. The first isomer leads to n-butyraldehyde, whereas the second isomer leads to isobutyraldehyde. The first isomer predominates under all conditions, but the isomeric ratio is dependent on the metal, the temperature, and the ligands. As the technology has improved, higher and higher ratios in favor of n-butyraldehyde have been obtained.
6.2.3 Hydrozirconation (Zr)
In hydroboration, a boron hydride adds across an alkene to give . The 16-electron, Zr(IV) complex , popularly known as Schwartz’ reagent, undergoes a closely related reaction.
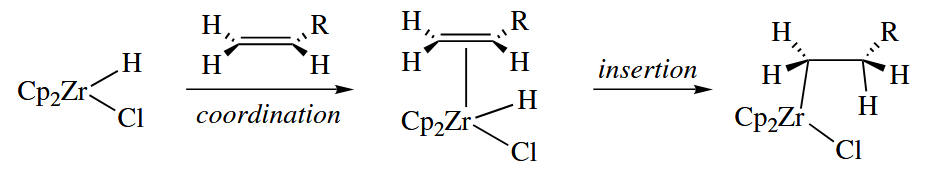
The alkylzirconium(IV) complexes produced by hydrozirconation are usually used as reagents in further transformations. The C-Zr bond is moderately nucleophilic at C, so hydrolysis cleaves the C-Zr bond and gives an alkane, whereas treatment with an electrophilic halogen gives an alkyl halide.
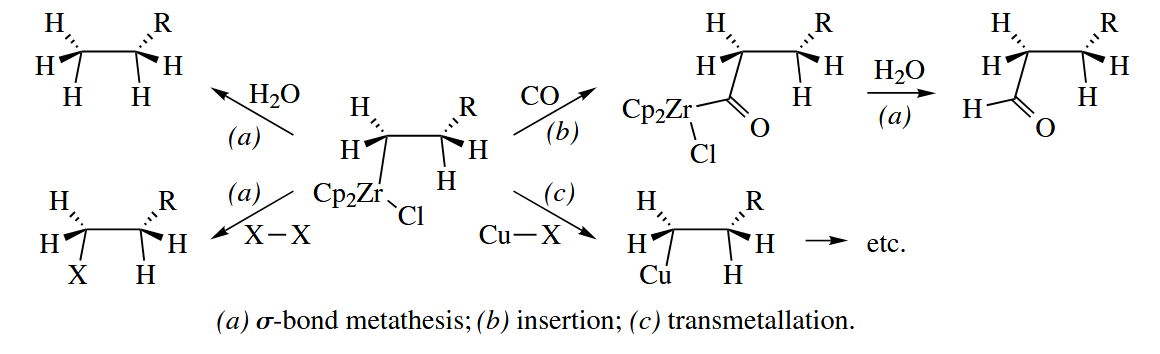
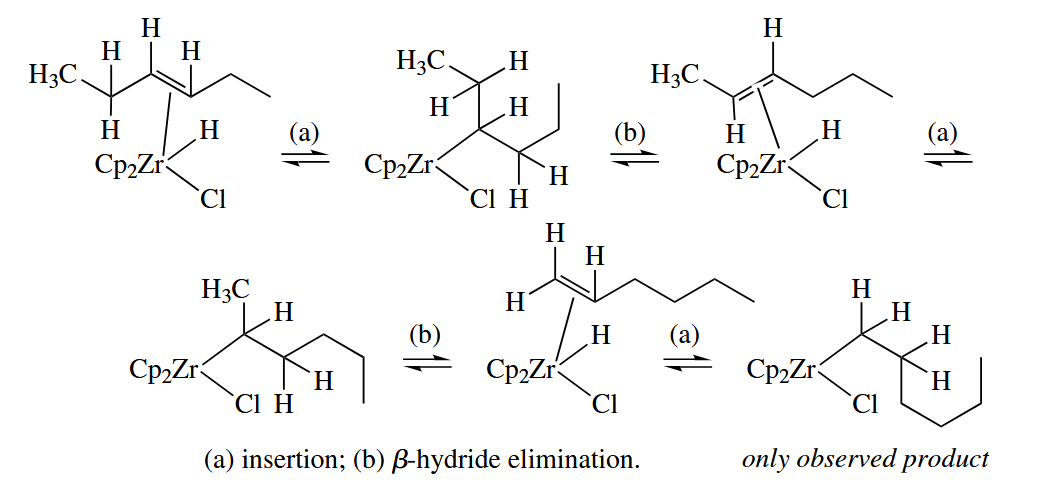
As mentioned earlier, hydrozirconation of internal alkenes gives terminal alkylzirconium compounds. The isomerization occurs so quickly that no intermediates are observed.
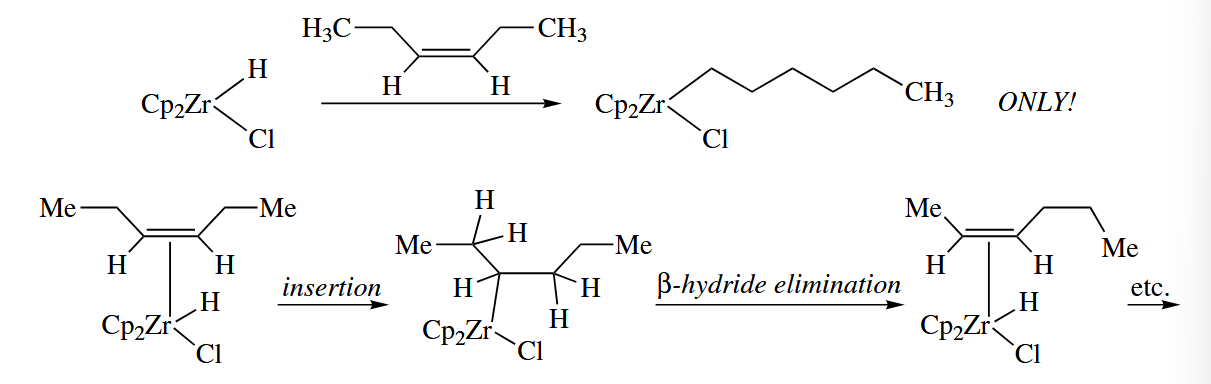
6.2.4 Alkene Polymerization (Ti, Zr, Sc, and Others)
The polymerization of ethelene to give polyethylene is one of the most important industrial reactions in the world. A free-radical process is most commonly used to execute the polymerization of ethylene.
The polymerization of higher alkenes such as propene, styrene, and butadiene is instead usually carried out using early-metal catalysts called Ziegler-Natta catalysts.
The most widely studied homogeneous Ziegler-Natta catalysts are the group 4 metallocenes, especially zirconocene. The catalytic cycle for alkene polymerization, the Cossee mechanism, is extremly simple. Coordination of the alkene to is followed by insertion to give a new complex . No change in the oxidation state of Zr occurs in either of these steps.
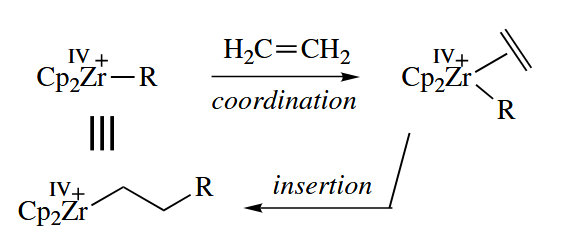
These are several mechanisms for termination of the growing polmer chain.
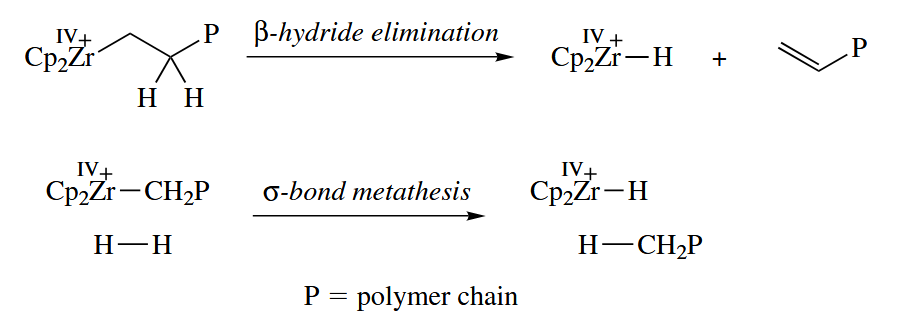
An alternative mechanism for alkene polymerization, the Green mechanism, has been demonstrated for the 12-electron Ta(III) complex .

The chain termination steps are the same as for the Cossee mechanism. The Green mechanism requires an increase in the oxidation state of the metal, and for this reason metals cannot polymerize alkenes by the Green mechanism.
( 上述机理又称 Green-Rooney 机理,由 Green 和 Rooney 两人于1978年提出。然而 Grubbs 等人于1985年发文指出了该机理的问题,他们通过同位素动力学实验证明:在烯烃聚合反应中,α-H 活化作用并没有那么强,因而证明 Green-Rooney 机理是不正确的。更多细节可见:Grubbs et al., J. Am. Chem. Soc., 1985, 3377. Brookhart 等人在1983年提出了 Brookhart-Green 机理,基本上是 Cossee 机理的补充,考虑了较微弱的 α-agostic 作用对烯烃 1,2-插入的影响。 )
When alkenes higher than ethylene are polymerized, regio- and stereochemical issues arise.
6.2.5 Cyclopropanation, Epoxidation, and Aziridination of Alkenes (Cu, Rh, Mn, Ti)
You may remember that the formation of carbenoids from diazo compounds such as is catlyzed by Cu(II) and Rh(II) complexes and that these carbenoids undergo [ 2 + 1 ] cycloadditions with alkenes as if they were singlet carbenes.
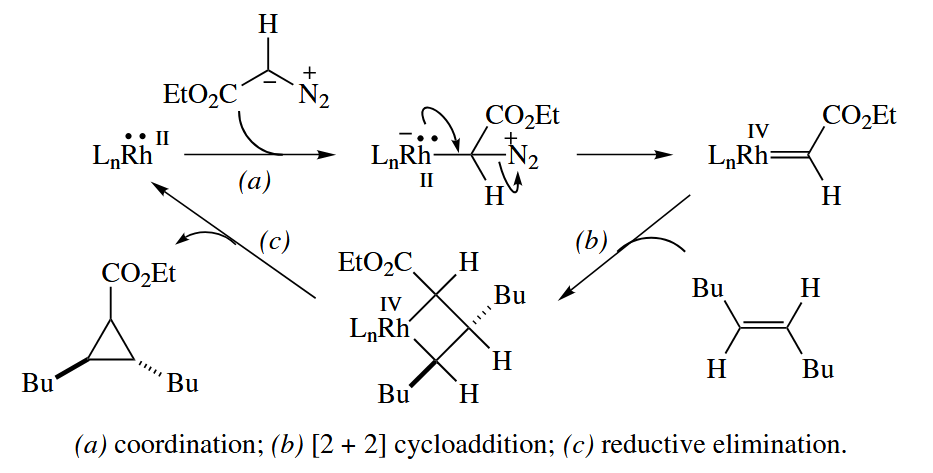
A number of other one-atom transfer reactions are mechanistically similar to the Rh- and Cu-catalyzed cyclopropanations.
For example, chiral Mn(II) salen complexes are widely used to catalyze the Jacobsen-Katsuki epoxidation of alkenes.
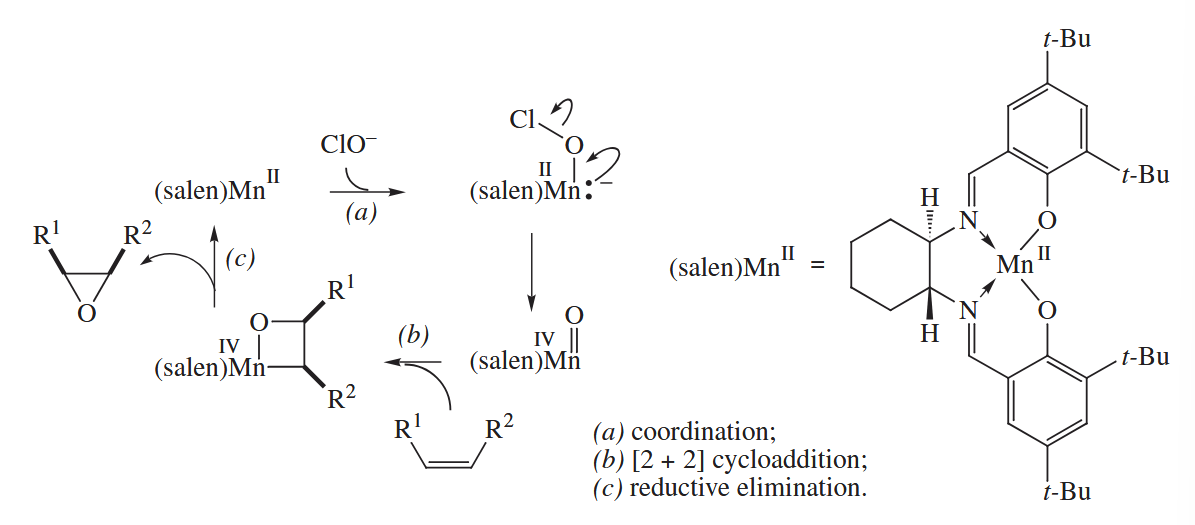
( 反应的氧化剂除了使用漂白粉外也可以用有机碘氧化物。 )
Ugly facts, unfortunately, sometimes invalidate a beautiful mechanism. The Jacobsen epoxidation sometimes proceeds with loss of configurational purity of acyclic alkenes. This feature of the reaction can be explained by invoking radicals.
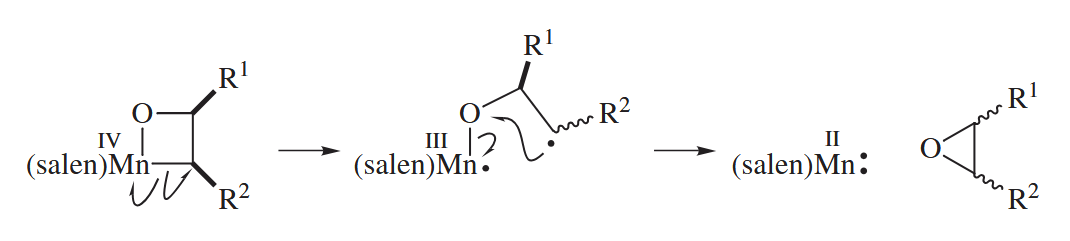
( 反应的对映选择性取决于很多因素,包括烯烃的结构、配体的性质和反应的温度等,反应对于顺式的1,2-二取代的烯烃选择性很好,但是反式的1,2-二取代烯烃则不尽人意。
另一点是本书中的催化剂写的是 (salen)Mn(II),但是通常认为是 (salen)Mn(III) 参与催化,其中 Mn 的氧化态在 III 到 V 之间转化。 )
The O atom source can be replaced with a source of NR such as PhI=NTs, and in this case, an aziridination reaction is catalyzed. The catalyst is usually a Cu(II) complex.
The sharpless epoxidation proceeds by a completely different mechanism from these reactions. This asymmetric O-transfer reaction uses catalytic amounts of and (+)- or (-)-diethyl tartrate to catalyze the epoxidation of allylic alcohols by t-BuOOH. The enantioselectivities are usually very good.
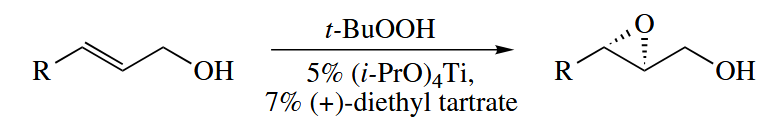
The metal acts essentially as a Lewis acid in this reaction. The terminal O atom is transferred selectively to one enantioface of the nucleophilic C=C π bond.
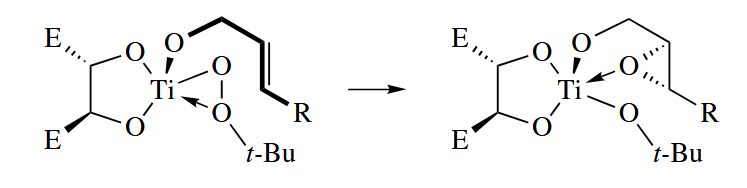
6.2.6 Dihydroxylation and Aminohydroxylation of Alkenes (Os)
When an alkene is treated with , an osmate ester containing two new C-O bonds is formed. The osmate ester is hydrolyzed, usually with aqueous , to give a 1,2-diol. The overall dihydroxylation reactions is stereospecifically syn.
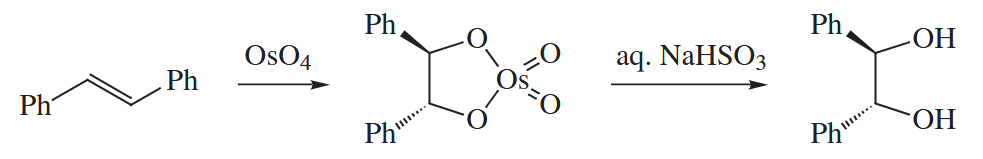
These are two major contenders for the mechanism of this reaction. Which mechanism is in fact operative remains controversial.
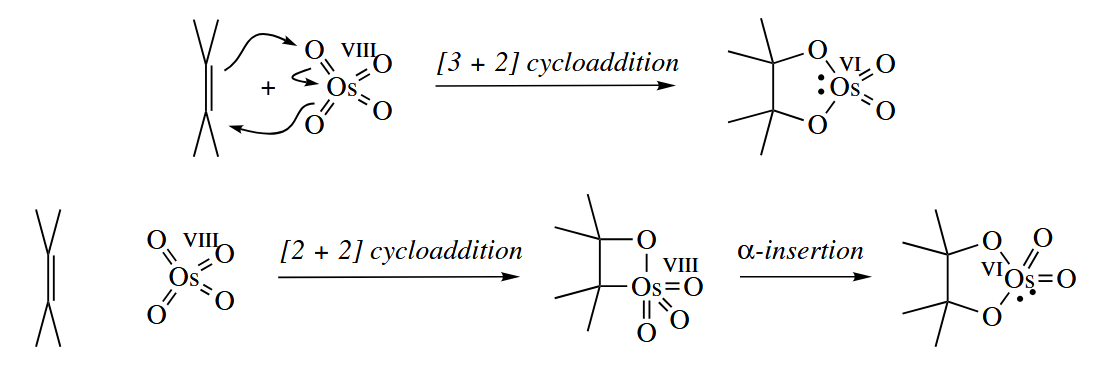
can catalyze the dihydroxylation of alkenes in the presence of a stoichiometric oxidant and water. The catalytic reaction is valuable because is very expensive and extremely toxic.
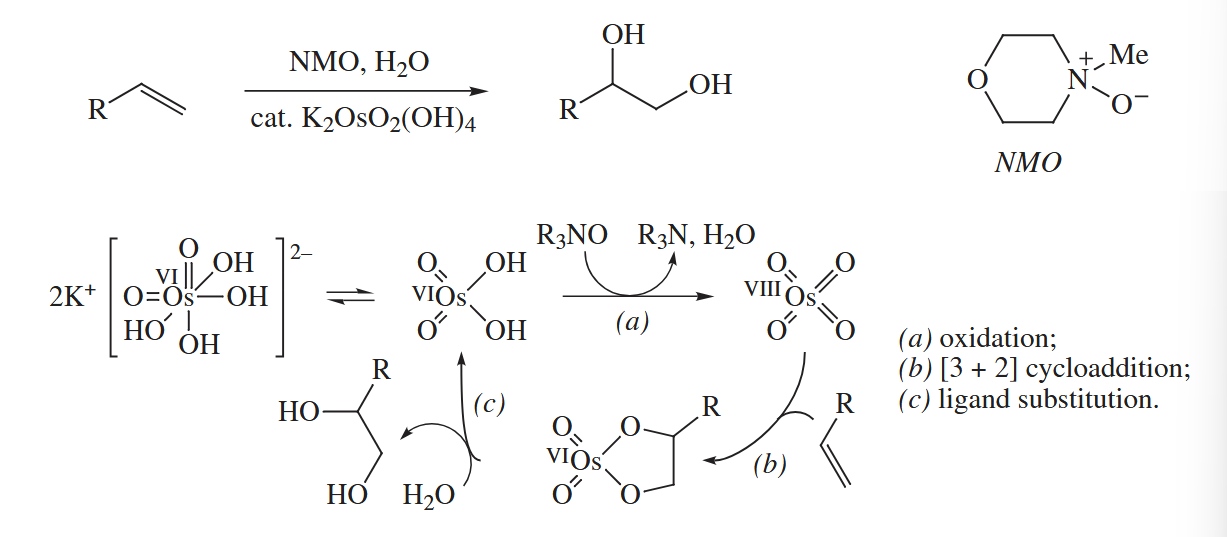
-mediated dihydroxylation is greatly accelerated by amines. When an alkene is combined with a catalytic amount of a chiral amine, a catalytic amount of or , and stoichiometric amounts of an oxidant [usually NMO or ] and , catalytic asymmetric or Sharpless dihydroxylation occurs. The combination is also used to catalyze the Sharpless asymmetric aminohydroxylation.
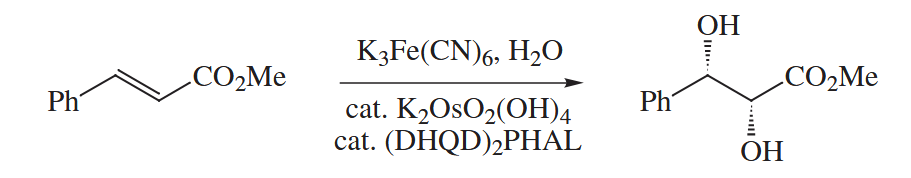
6.2.7 Nucleophilic Addition to Alkenes and Alkynes (Hg, Pd)
In the presence of Hg(II) salts such as , nucleophiles such as add readily to alkenes to give organomercury compounds. The C-Hg bond is readily converted to a C-H bond by the addition of .
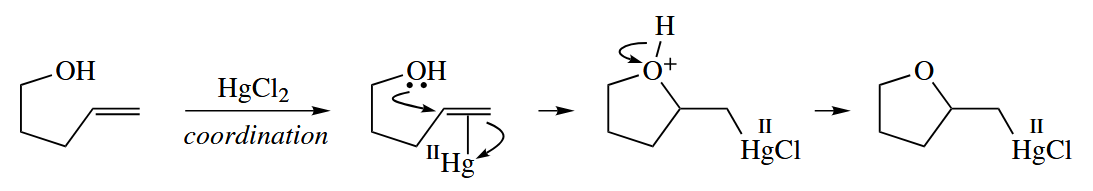
The converts RHgCl to RHgH by nucleophilic substitution. RHgH then undergoes free-radical decomposition to give RH and Hg(0).
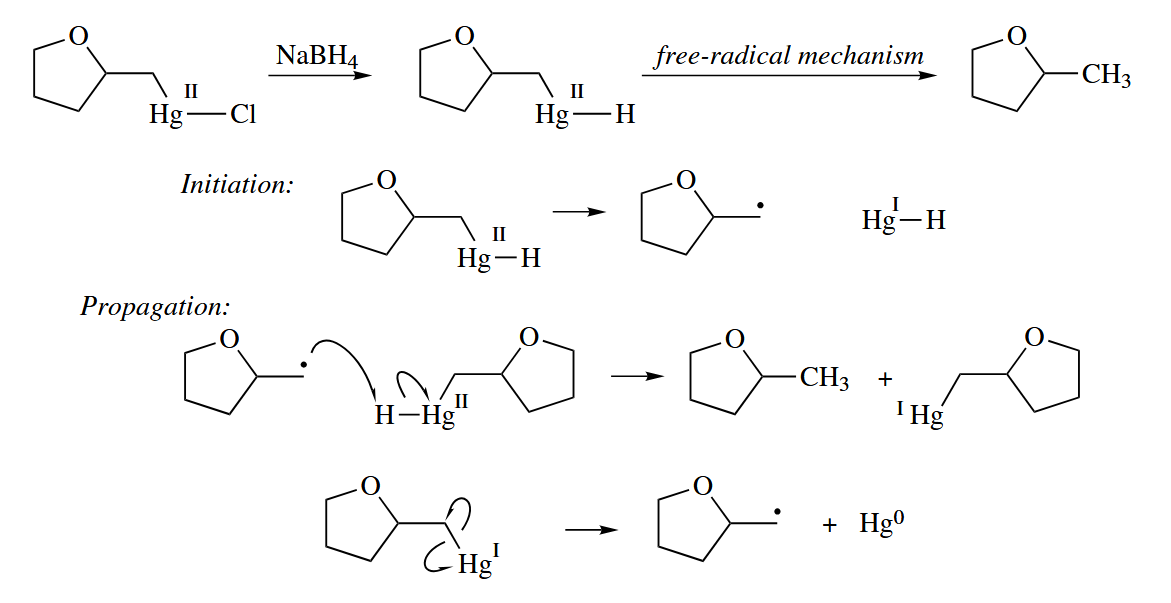
( 根据机理,反应最后会产生汞单质,事实上也的确如此,反应烧瓶的底部会出现小球状的汞珠,因此许多汞试剂已经被更高效、低毒性的试剂取代。 )
The addition of to alkynes is catalyzed by Hg(II) under acidic conditions.
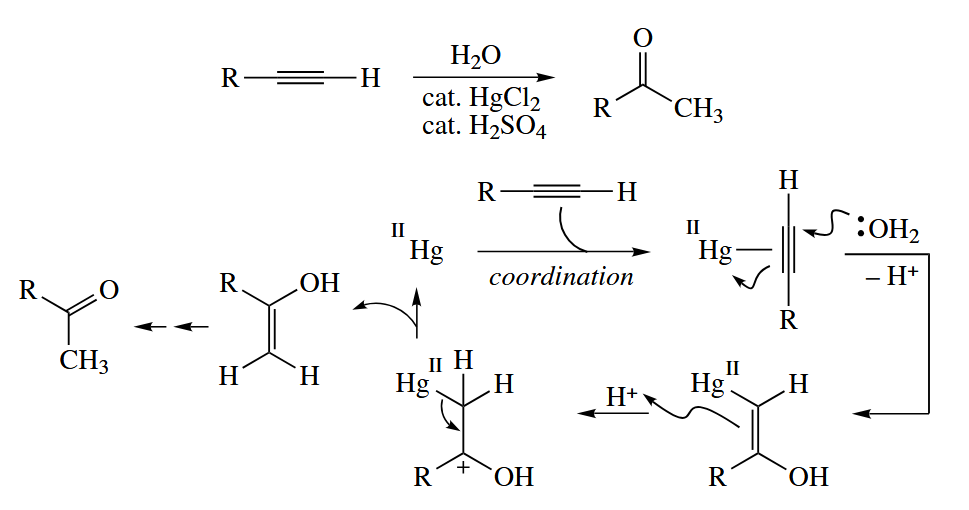
( 注意上图中在 coordination 的下一步,水分子对 Hg 活化的炔烃的进攻箭头绘制有误,应该符合马氏加成,当然后续步骤是没问题的。 )
Palladium salts also promote the addition of nucleophiles to alkenes and alkynes.
6.2.8 Conjugate Addition Reactions (Cu)
Compounds containing a C-Cu bond undergo conjugate addition to α,β-unsaturated carbonyl compounds, especially ketones.
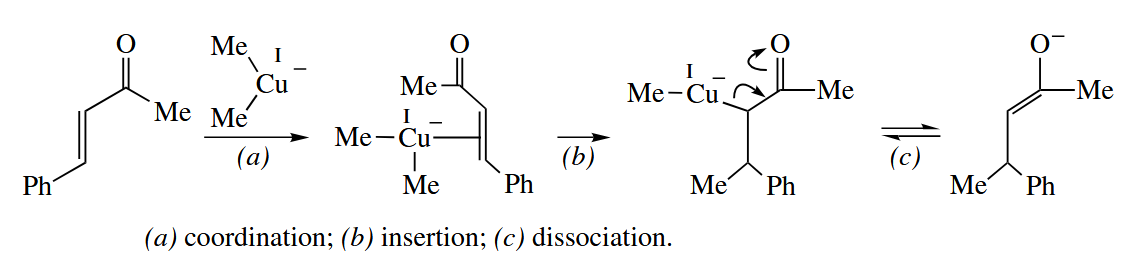
In the presence of a catalytic amount of a Cu(I) salt, Grignard reagents add exclusively to the β-carbon of α,β-unsaturated carbonyl compounds. The reaction proceeds via transmetallation of the Grignard reagent (RMgX) with CuX to give either RCu or . The alkylcopper compound undergoes conjugate addition to the enone. Transmetallation of the Cu enolate with RMgX gives the Mg enolate and complete the catalytic cycle.
6.2.9 Reductive Coupling Reactions (Ti, Zr)
The group 4 metals, especially Ti and Zr, are most stable in their (IV) oxidation states. In their (II) oxidation states they act as reducing agents.
The reductive coupling of two carbonyl compounds using reduced Ti reagents is called McMurry coupling. The products may be 1,2-diols or alkenes.
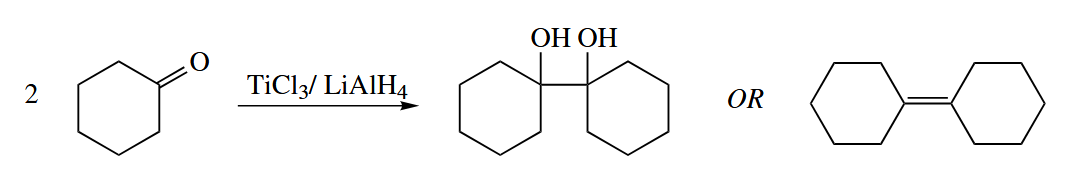
The mechanism is obscure. One can imagine that the reduced Ti(II) forms a π complex with one carbonyl compound to give a Ti(IV) titanaoxirane.
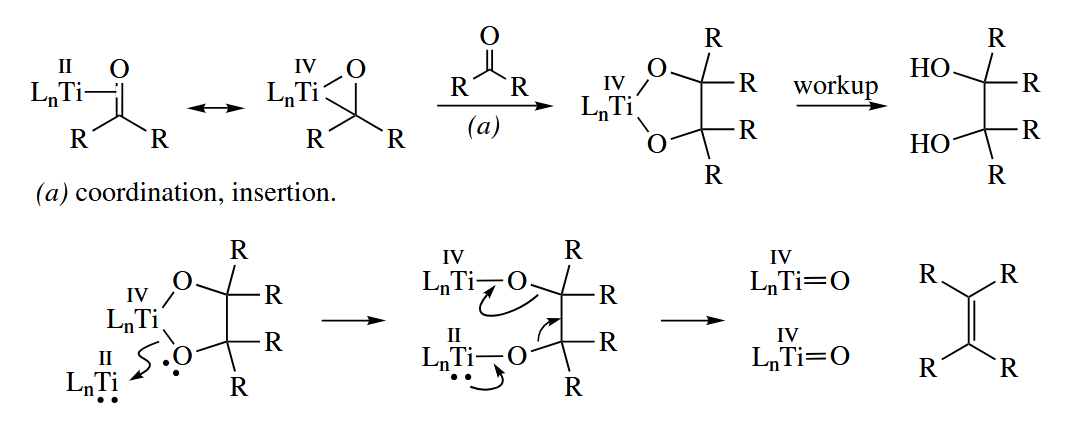
In another mechanism for McMurry coupling, a Ti(IV) ketyl forms from Ti(III) and the carbonyl. Radical-radical combination then gives the 1,2-diol in a pinacol coupling.
( 文中所述的 “another mechanism” 其实相对更常见一些,即通过 SET 机理实现的类似 pinacol coupling。 )
Reductive coupling of alkynes or alkenes may be carried out with Ti(IV) and Zr(IV) complexes. The reaction is often carried out in an intramolecular fashion to achieve better control of regiochemistry.
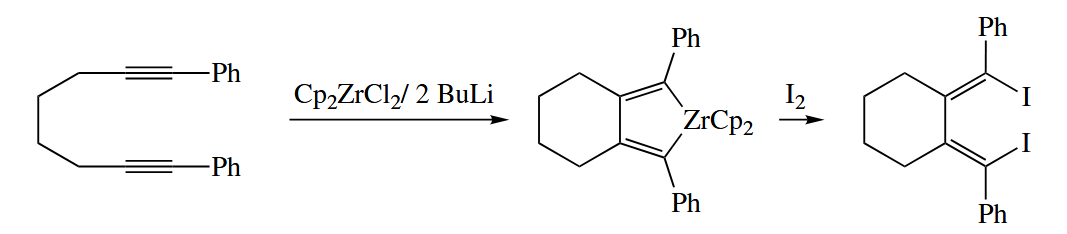
( 上图反应中 BuLi 的作用是还原 Zr(IV) 到 Zr(II),除此之外用金属作还原剂也是非常常见的,例如 McMurry Coupling 常用 Zn 作还原剂。 )
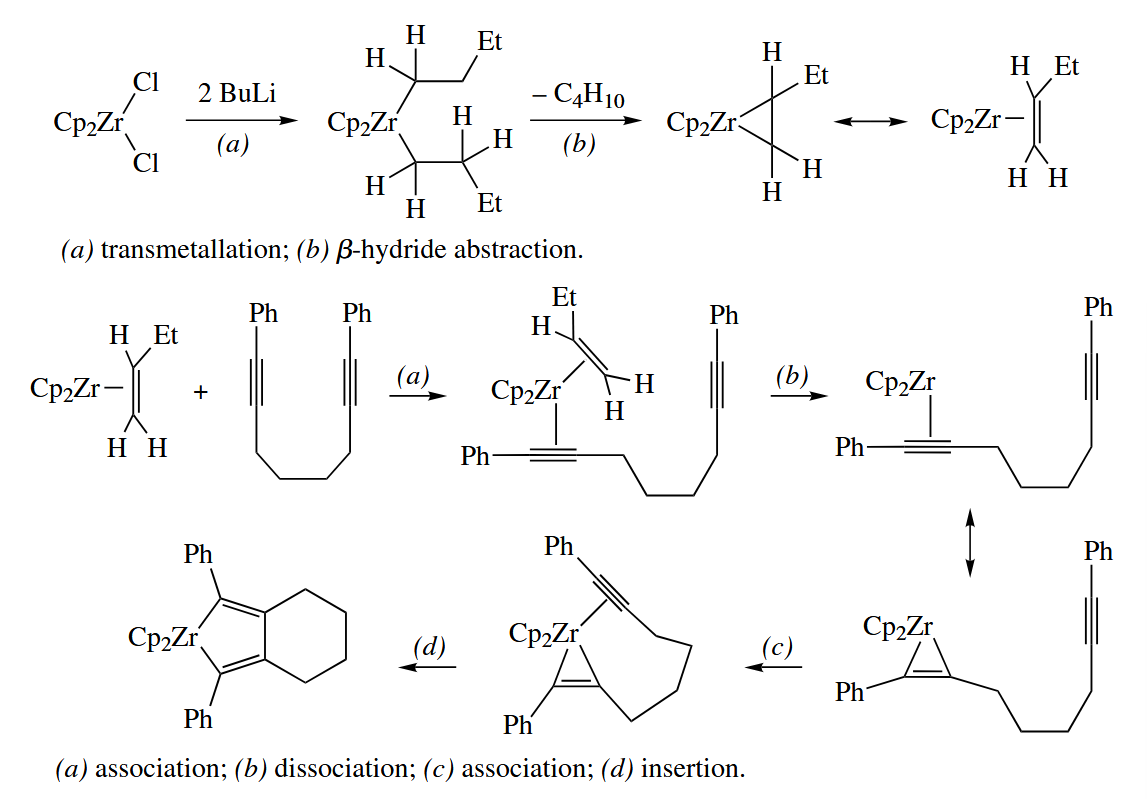
When an enyne is reductively coupled in an intramolecular fashion with Zr or Ti, the metallacyclopentene that is obtained initially may be carbonylated to the corresponding bicyclic cyclopentenone with CO.
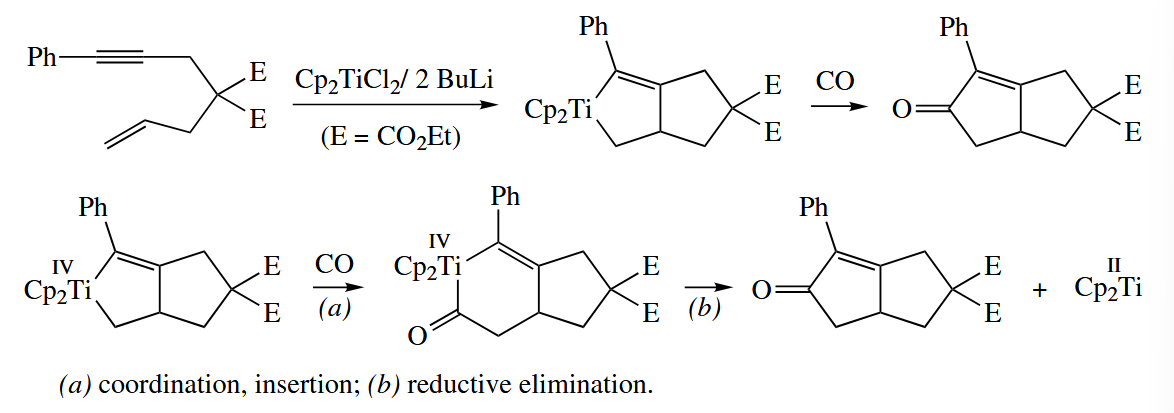
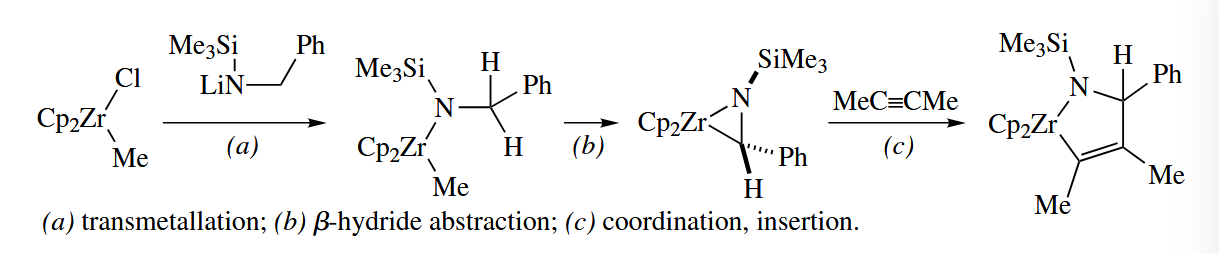
A wide variety of π complexes , including complexes of alkynes, cycloalkynes, arynes, imines, and thioaldehydes, can be generated by β-hydride abstraction from complexes of the type .
The Kulinkovich cyclopropanation, in which an ester and two equivalents of a Grignard reagent are combined to make a cyclopropanol in the presence of a catalytic amount of .
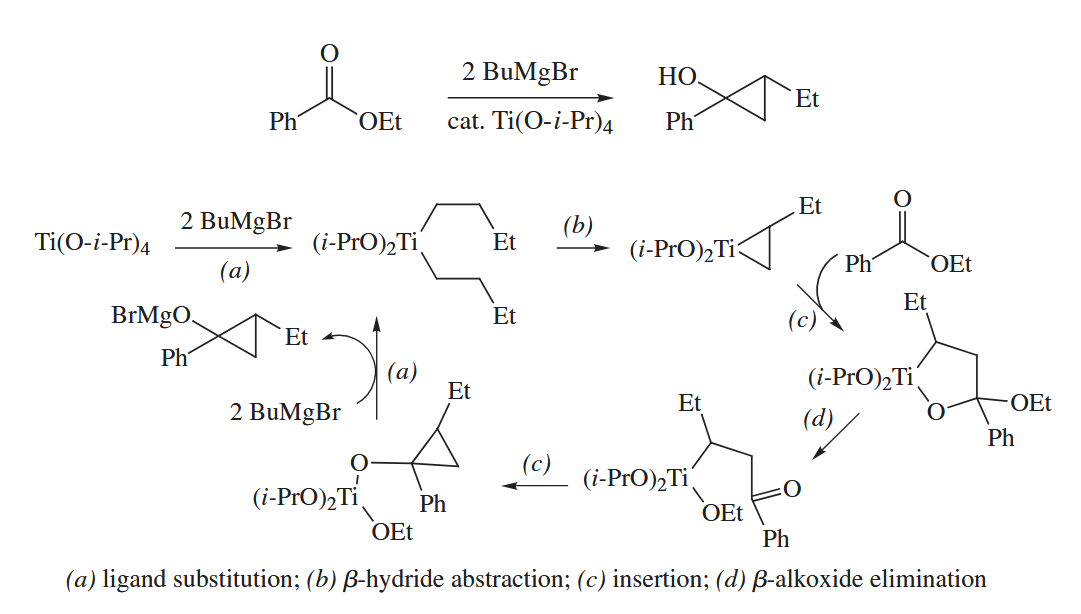
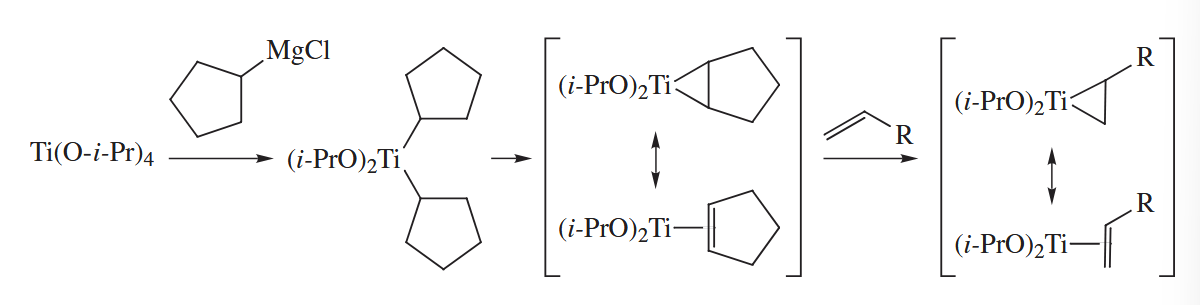
When an external alkene is added to the reaction mixture, ligand exchange can generate a new alkene-titanium complex into which the ester can insert. Thus, two equivalents of an inexpensive cheap Grignard reagent can be sacrificed to couple a precious alkene to the ester or amide.
6.2.10 Pauson-Khand Reaction (Co)
The Pauson-Khand reaction combines an alkyne, an alkene, and CO to give a cyclopentenone.
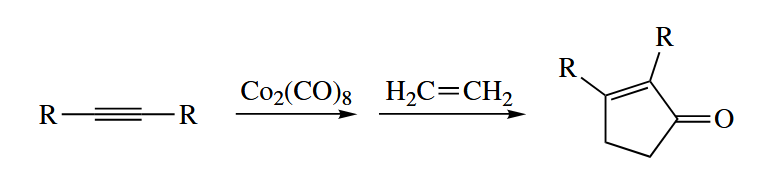
When an alkyne is added to , CO evolves, and an isolable, chromatogaphable “butterfly” alkyne- complex is obtained.
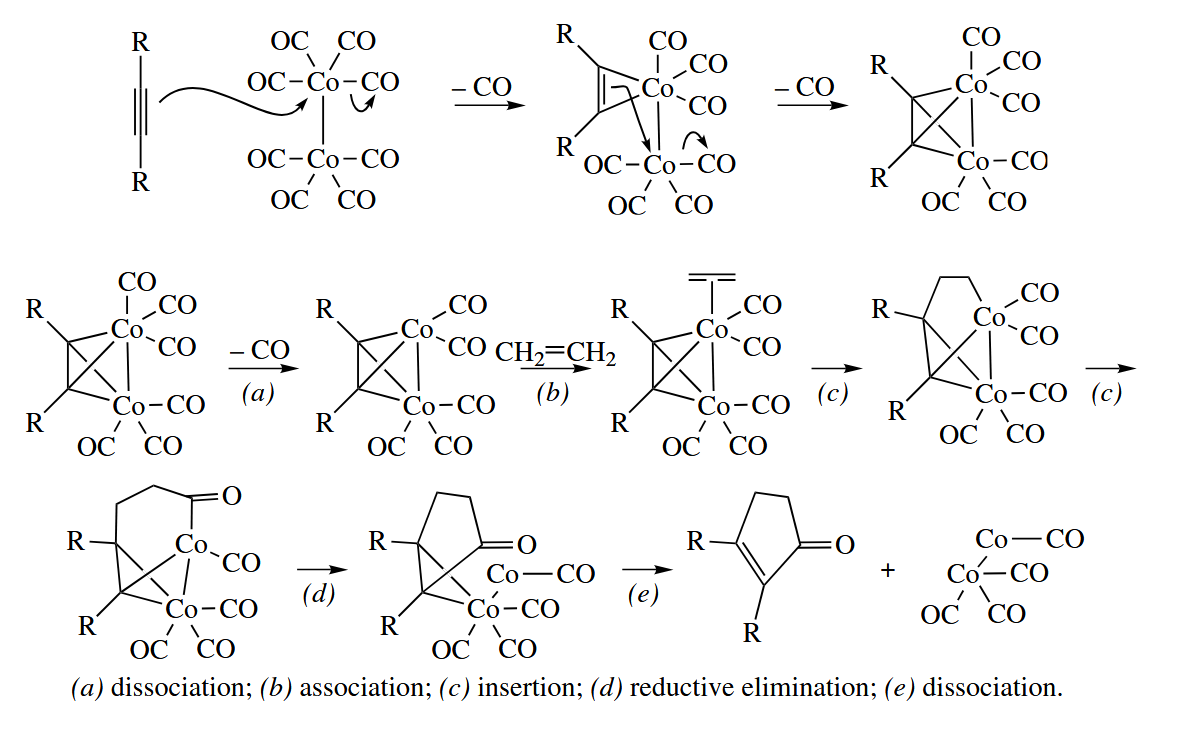
The Pauson-Khand reaction is especially useful for the intramolecular cyclization of 1,6-enynes to give bicyclo[3.3.0]octenones.
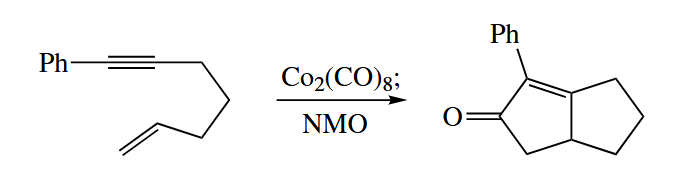
6.2.11 Dötz Reaction (Cr)
In the Dötz reaction, an unsaturated chromium carbene complex is combined with an alkyne to give a substituted phenol. There is almost always a methoxy or other alkoxy group attached to the carbene.
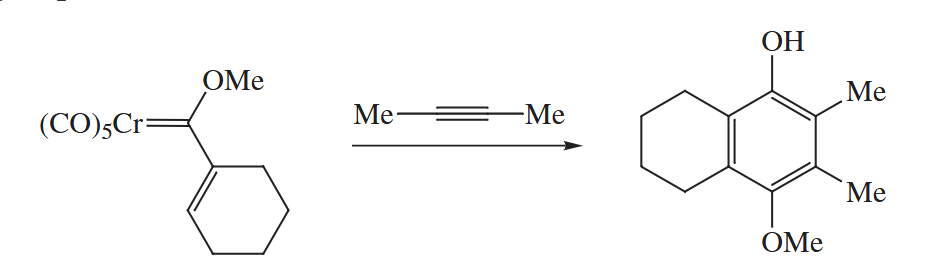
By numbering the atoms, we see that the alkylidene portion of the Cr complex contributes three C atoms to the new aromatic ring, and the alkyne contributes two. The sixth C atom with its associated O must come from a CO ligand.
Compounds with multiple M-C bonds are prone to undergo [ 2 + 2 ] cycloadditions. Then incorporation of the CO group into the ring and formation of the two remaining the C-C bonds can be formulated.
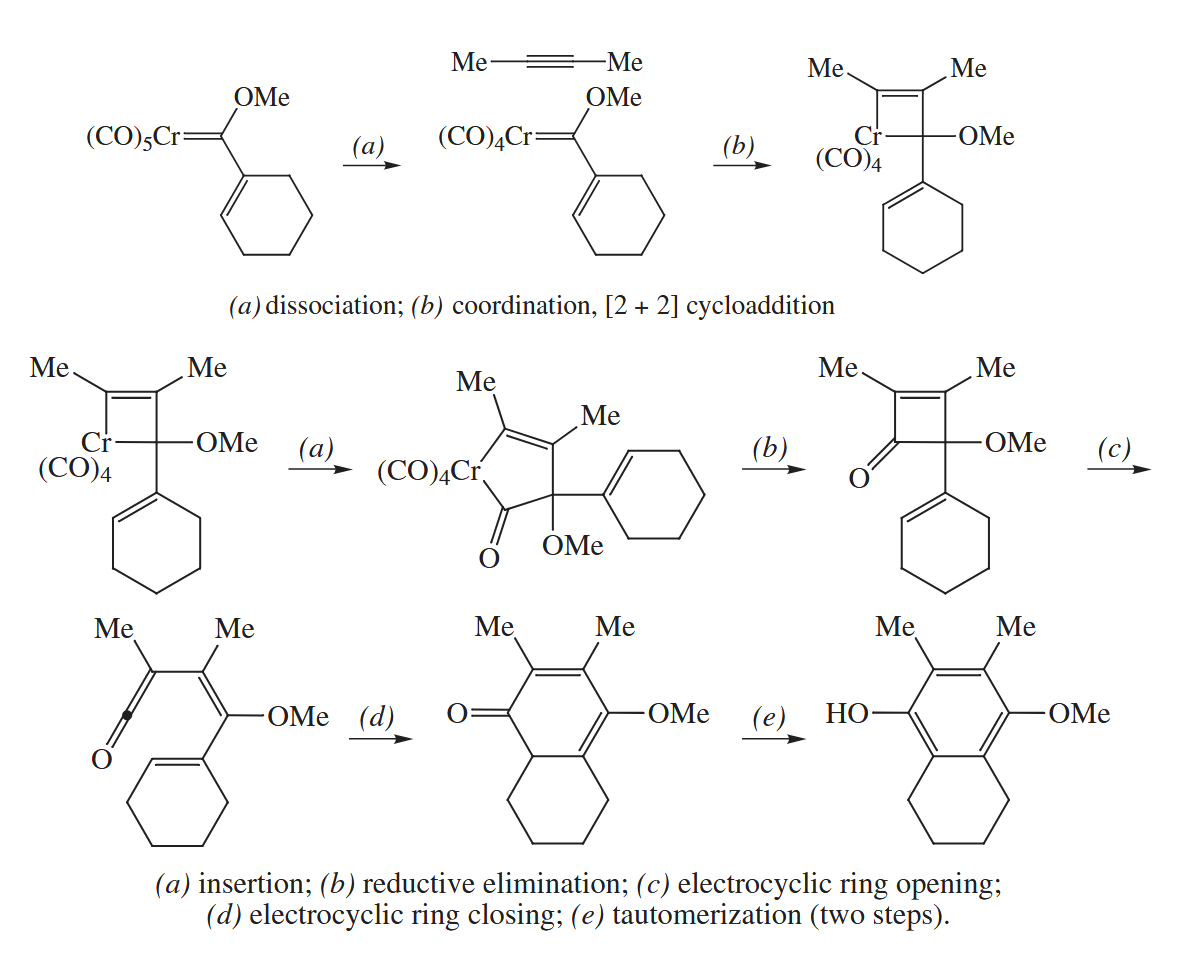
( 该反应的机理具有多种可能,我仅列出了相对多见的一种。 )
The Cr-containing starting material for the Dötz reaction is usually prepared from . The CO groups attached to Cr in this compound exert such a strong electron-withdrawing effect on the Cr=C bond. In fact, the fragment is so electron-attracting that the group becomes as acidic as a methyl ketone!
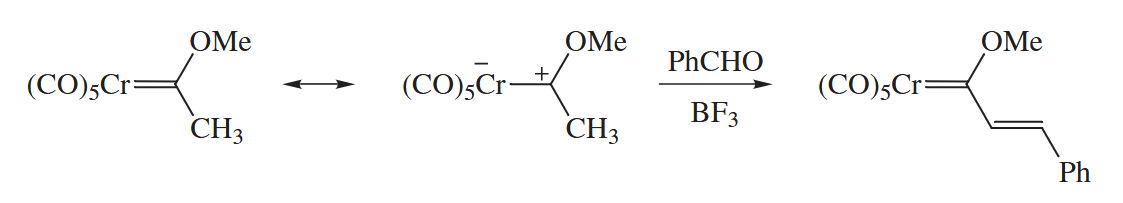
6.2.12 Metal-Catalyzed Cycloaddition and Cyclotrimerization (Co, Ni, Rh)
Certain late-transition-metal complexes have the ability to catalyze [ 4 + 2 ], [ 4 + 4 ], and [ 5 + 2 ] cycloadditions. Despite their appearance, these reactions are not concerted cycloadditions.
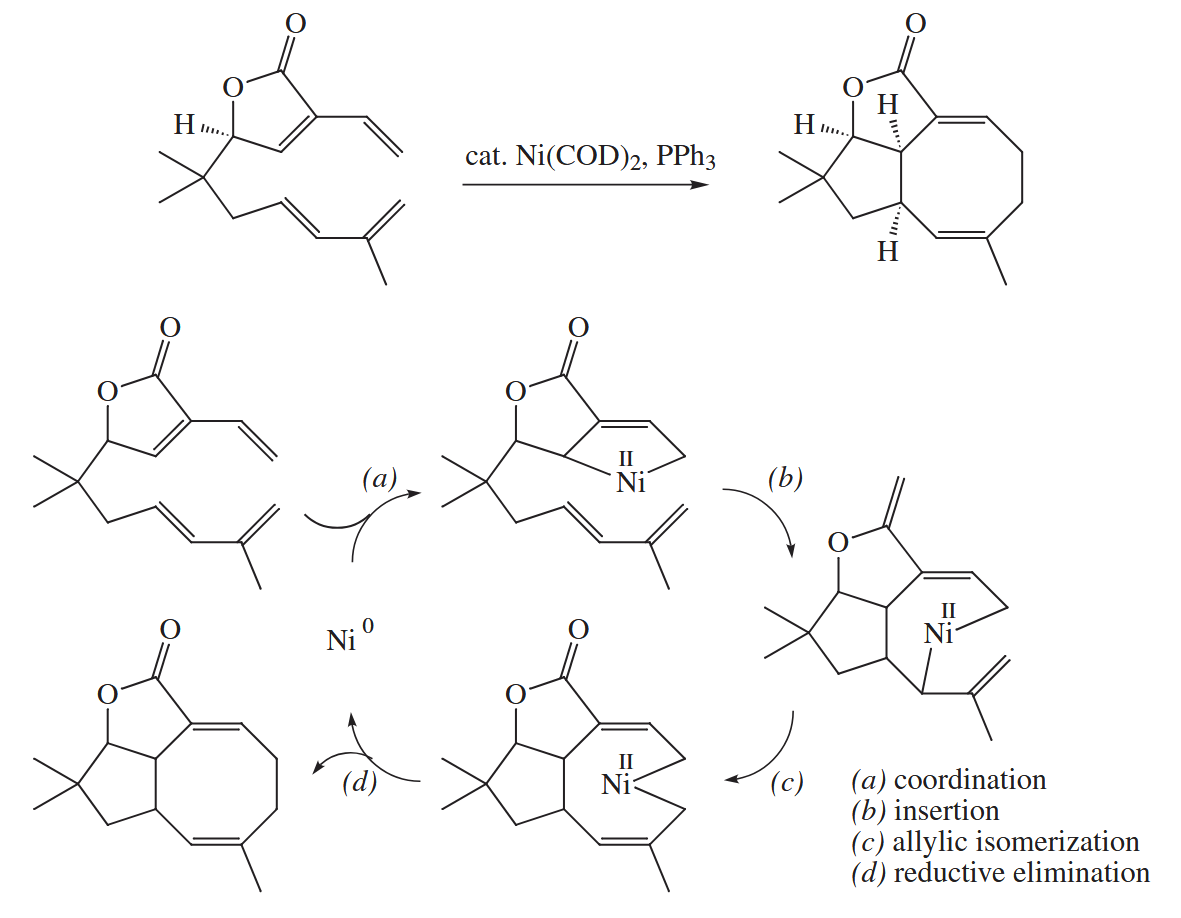
The Rh-catalyzed [ 5 + 2 ] cycloaddition combines a vinylcyclopropane and an alkyne (or an alkene or allene) to give a cycloheptadiene.
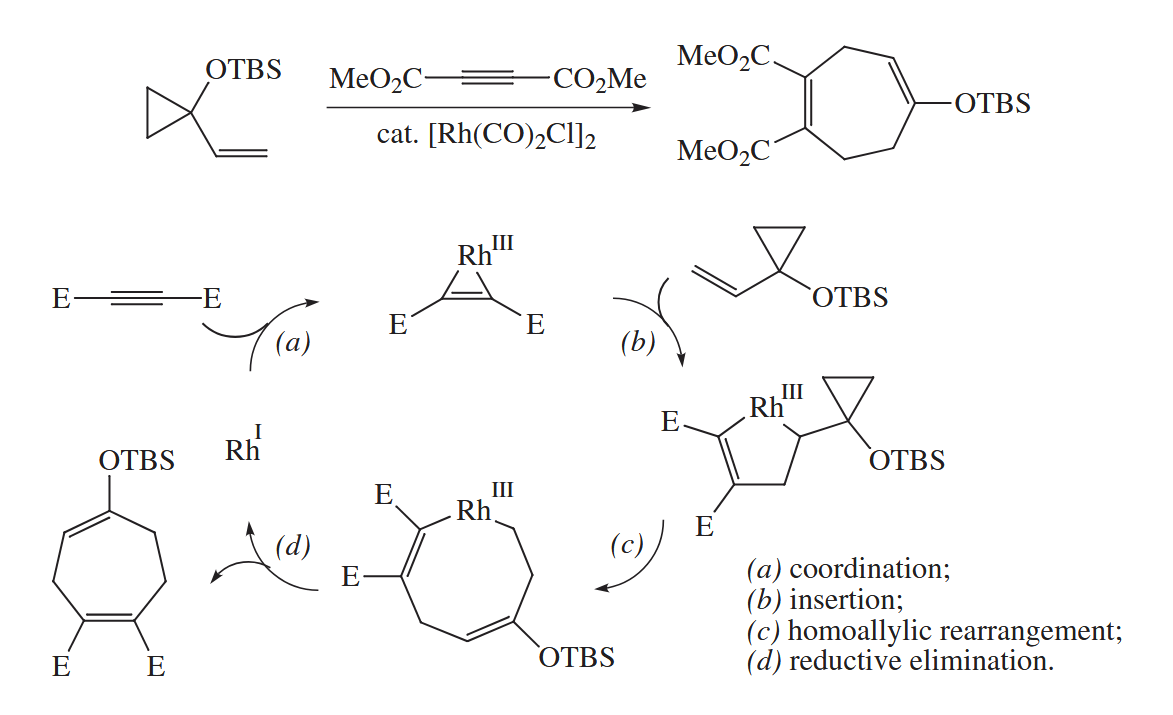
( 环丙烷结构在很多时候的反应性都和烯烃类似,此处是通过均裂的烯丙基重排完成的环化,表观上和迁移插入的结果是一致的。 )
Various Ni and Co complexes catalyze cyclotrimerization reactions of alkynes to give arenes. These reactions are formally [ 2 + 2 + 2 ] cycloadditions.
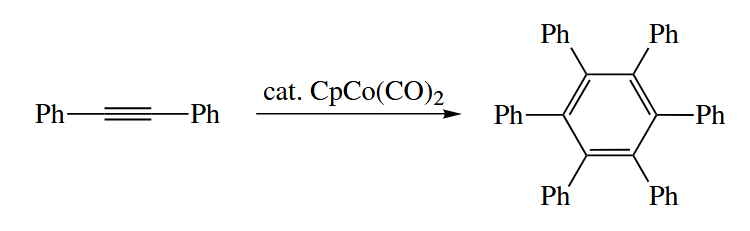
The cyclotrimerizaiton reaction has been used to prepare some remarkable compounds.
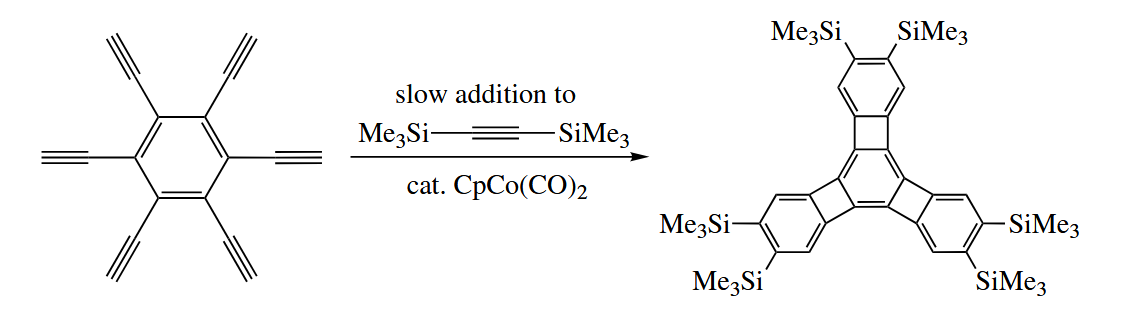
6.3 Substitution Reactions
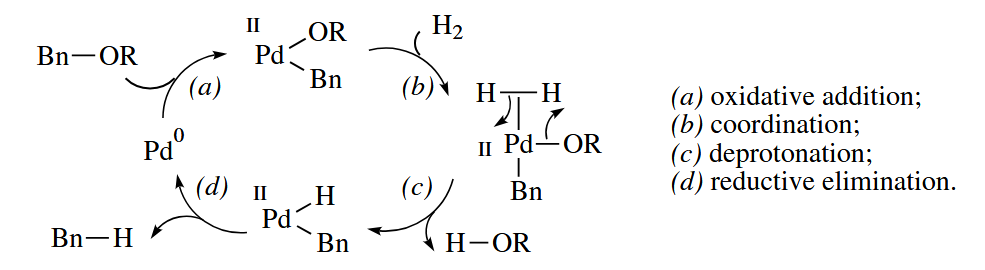
6.3.1 Hydrogenolysis (Pd)
Hydrogenolysis catalyzed by Pd/C is widely used to convert benzylic ethers to arenes. One reasonable catalytic cycle can be drawn.
6.3.2 Carbonylation of Alkyl Halides (Pd, Rh)
A Pd(0) catalyst such as catalyzes the alkoxycarbonylation of an organic halide (RX) to an ester () in basic MeOH under a CO atmosphere. (Any alcohol can be used.)
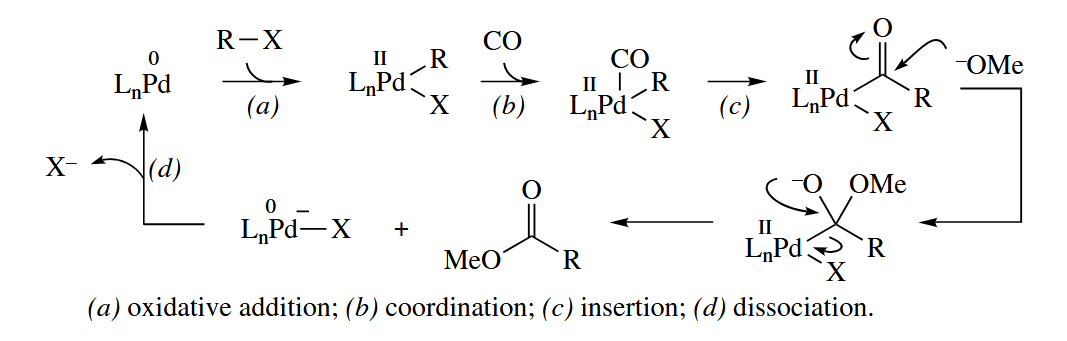
( 这个反应可以为我们提供一个通用的反应范式,许多 late metal 催化的取代反应机理往往相似,它们的第一步几乎都是氧化加成;金属的配体大多是大位阻的膦配体,较大的配体有利于金属在溶液中保持氧化态为 0 。 )
Sometimes, a Pd(II) compound is added to the reaction mixture as catalyst. The Pd(II) must be reduced to Pd(0) before the catalytic cycle can proceed.
The Pd-catalyzed carbonylation of alkyl halides is an extraordinarily useful reaction. The Pd-catalyzed reaction proceeds at room temperature or slightly higher temperatures and requires only a weak base like .
An important limitation to the reaction, however, is that R must almost always be C(). The only C() halides that can undergo the reaction are those lacking β-hydrogens.
The Monsanto process, one of the most successful industrial homogeneous catalytic processes, uses a Rh complex and catalytic HI to carbonylate MeOH to .
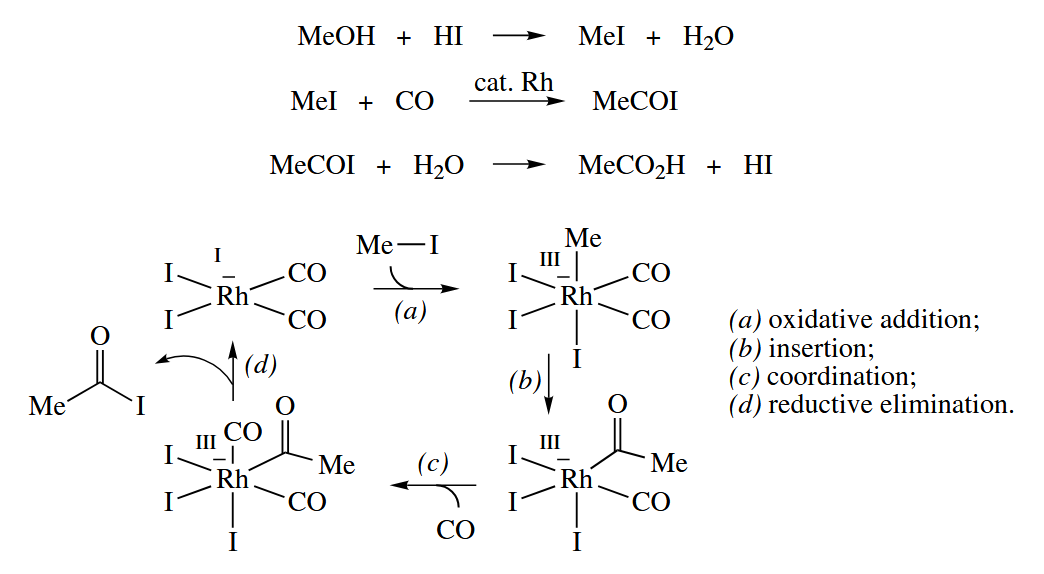
6.3.3 Heck Reaction (Pd)
In the Heck reaction, an aryl or vinyl halide (R-X) and an alkene are converted to a more highly substituted alkene under Pd catalysis.
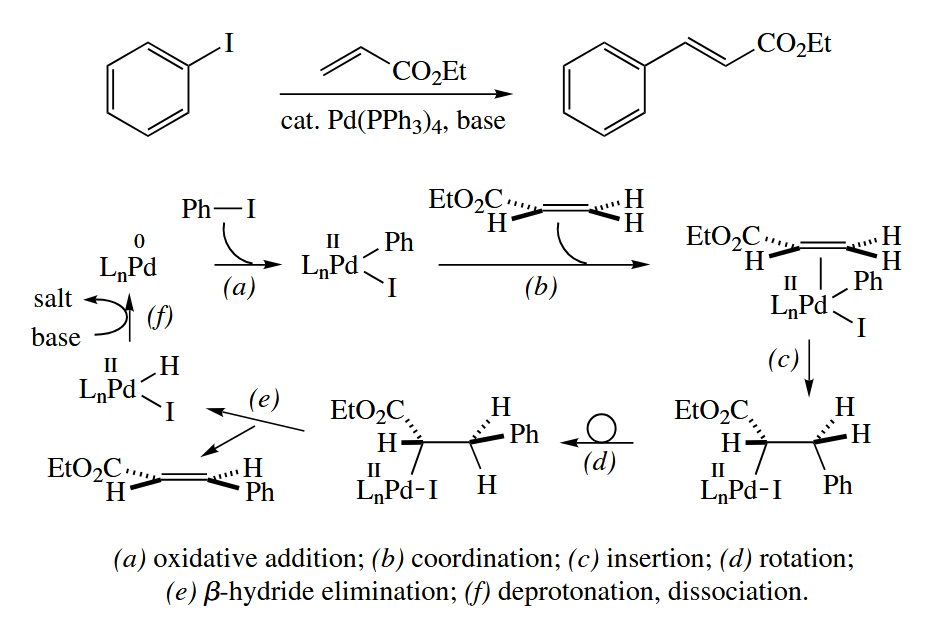
Primarily trans alkenes are obtained due to conformational preferences in the rotamer from which β-hydride elimination takes place. If the starting material is prochiral, the reaction can be made asymmetric by the use of chiral phosphine ligands like BINAP.
6.3.4 Coupling Reaction Between Nucleophiles and C()-X: Kumada, Stille, Suzuki, Negishi, Buchwald-Hartwig, Sonogashira, and Ullmann Reactions (Ni, Pd, Cu)
Aryl and vinyl halides can undergo substitution with nucleophiles by one of three mechanisms: addition-elimination, , or elimination-addition. Many aryl halide-nucleophile pairs fulfill none of these conditions, and consequently this transformation was for a long time one of the most difficult ones to accomplish.
In the mid-1970s, it was discovered that Ni complexes catalyzed the substitution of aryl halides with Grignard reagents at room temperature. Experimental evidence suggests that the mechanism of the Kumada coupling is proposed to be as follows.
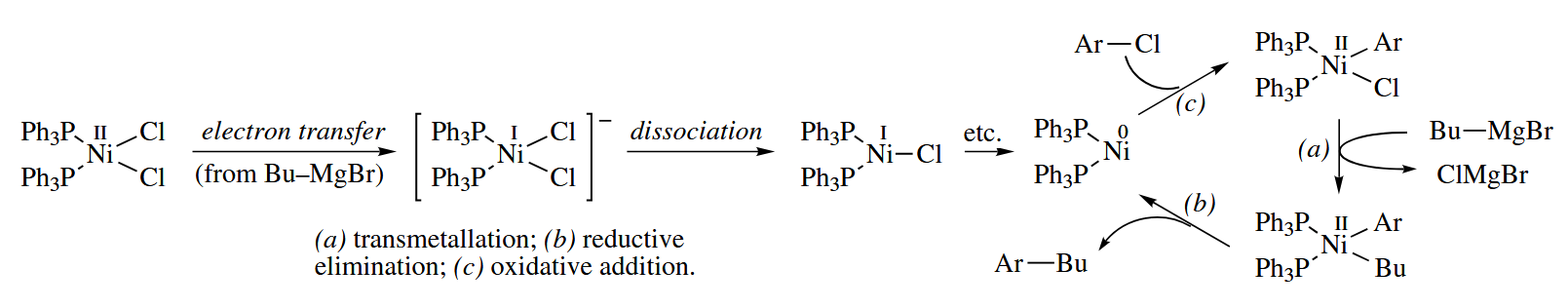
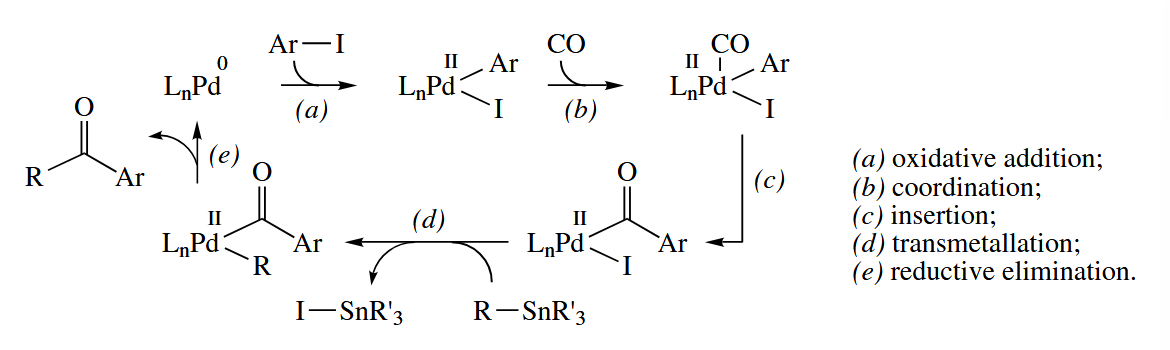
After the discovery of the Kumada reaction, intensive efforts were made to discover other group 10 metal-catalyzed reactions. These efforts paid off tremendously with the development of some of the most widely used C-C bond-forming reactions in organic synthetic methodology, including the Stille coupling, the Suzuki coupling, the Negishi coupling, and related reactions.
In these reactions, an aryl or vinyl halide or pseudohalide undergoes a Pd-catalyzed substitution reaction with a “nucleophilic” alkylmetal compound. The catalytic cycle, shown for the Stille coupling, involves oxidative addition to the Ar-X bond, transmetallation, and reductive elimination. The catalytic cycle is exactly the same for the Suzuki (), Negishi (), and other alkylmetal couplings.
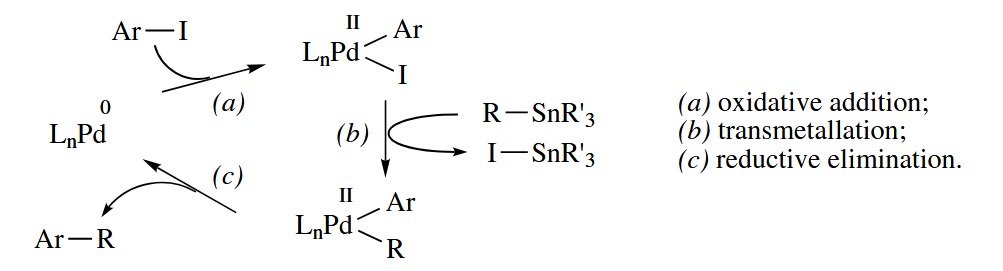
When the added catalyst is Pd(II), it is reduced to Pd(0) before the catalytic cycle begins. The reduction probably proceeds by two transmetallations from the nucleophile and a reductive elimination.
Metal amides and alkoxides are also coupled to C()-X electrophiles under Pd catalysis in Buchwald-Hartwig amination or etherification. The bulky phosphine ligands such as are required for these reactions to proceed. The ligand are thought to enforce a lower coordination number of the Pd catalyst, making a more active catalyst.
Many compouds containing metal-metal bonds undergo “Stille” and “Suzuki” couplings, too. In this case, the mechanism by which the B-H bond is converted to a B-Pd bond is somewhat murky.
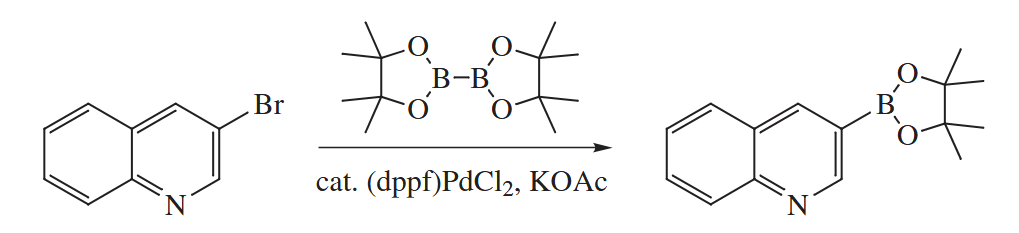
The carbonylative Stille coupling is another extremely useful reaction.
Terminal alkynes () also undergo Pd-catalyzed Sonogashira coupling to alkyl halides in the presence of base and a subcatalytic amount of CuI to give terminal alkynes ().
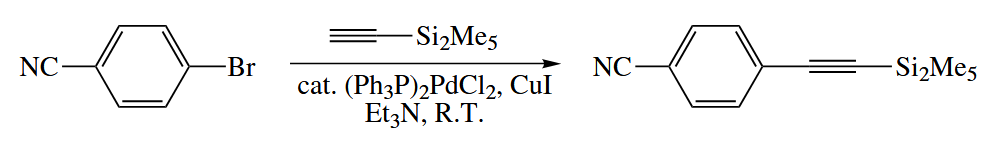
The CuI can cocatalyzes the reaction and lower the temperature required for the sonogashira coupling from >100℃ to room temperature.
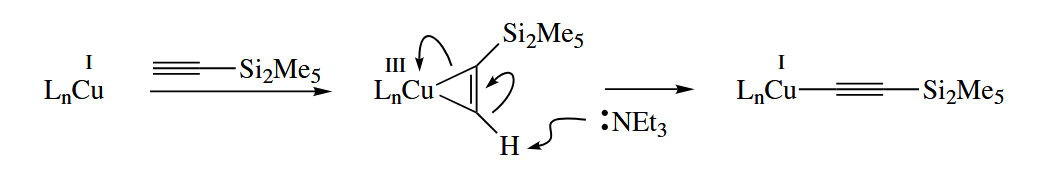
Copper(I) salts undergo aromatic substitution reactions very readily with ordinary aryl halides. The only problem with the oxidative addition-reductive elimination mechanism is that Cu(III) is a relatively high energy species. The mechanism would be much more reasonable if the Cu cycled between Cu(0) and Cu(II) instead of Cu(I) and Cu(III).
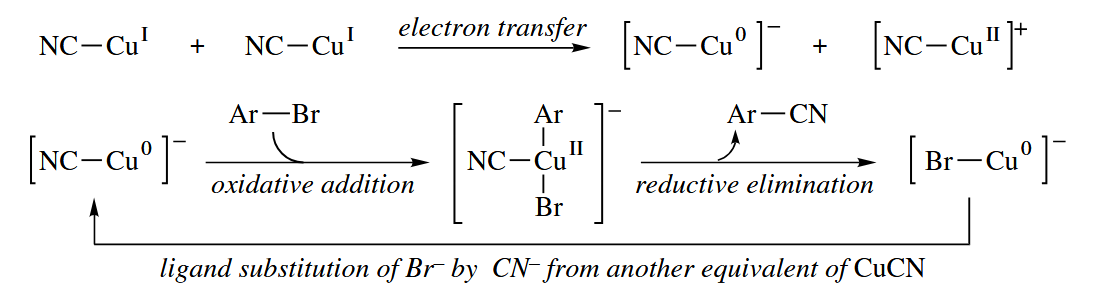
In the Ullmann reaction, Cu metal promotes the coupling of Ar-I to give Ar-Ar. The reaction mechanism almost certainly involves oxidative addition of Cu(0) to Ar-I to give Ar-Cu(II), then reduction by another equivalent of Cu(0) to give Ar-Cu(I).
6.3.5 Allylic Substitution (Pd)
In the presence of a Pd catalyst, allylic carbonates and acetates are substrates for the substitution reaction. Significantly, the Pd-catalyzed reaction occurs with retention of stereochemistry. In fact, double inversion occurs in this reaction.
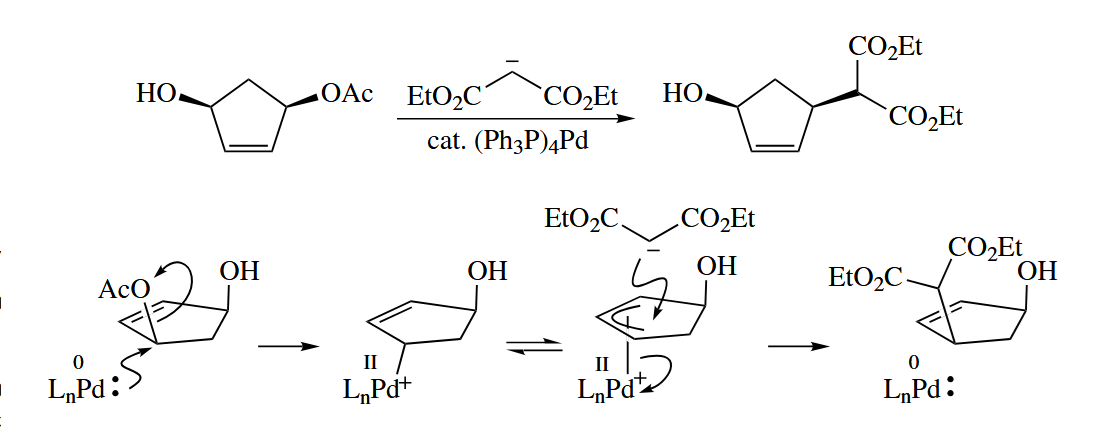
The Pd catalyst may be any of the commonly used Pd(0) or Pd(II) species. Pd complexes with chiral phosphine ligands can effect asymmetric allylations. The reaction works for allylic epoxides, too.
6.3.6 Palladium-Catalyzed Nucleophilic Substitution of Alkenes; Wacker Oxidation
Pd(II) salts catalyze the reaction of nucleophiles such as alcohols and amines with alkenes to give less substituted alkenes. A stoichiometric amount of an oxidant is required for the reaction to be catalytic in Pd.
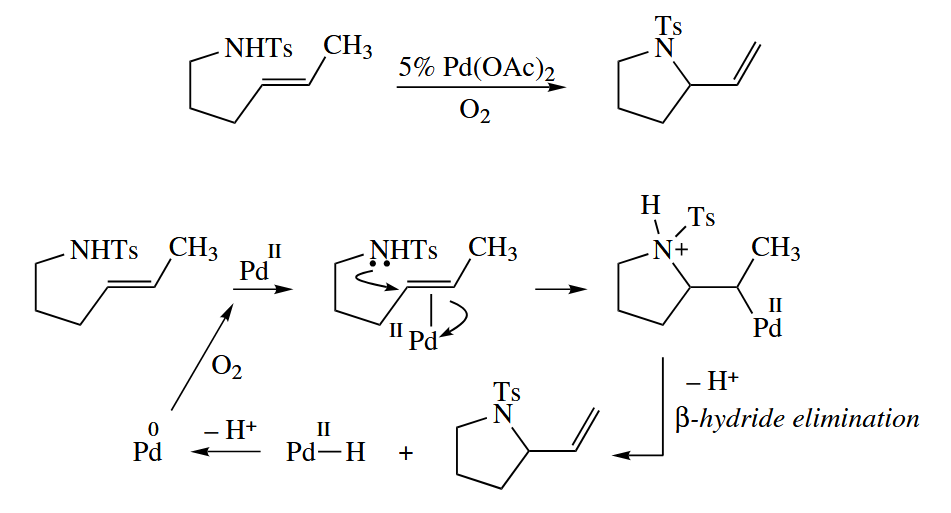
( 注意 β-氢消除反应速率快,反应可逆,因此即使多取代烯烃是热力学稳定的,也会因为可逆反应而得到端基金属烷烃,从而最终得到少取代的烯烃。 )
The Wacker oxidation is used industrially to convert ethylene and into acetaldehyde. The O atom in the product comes from the water, not the .
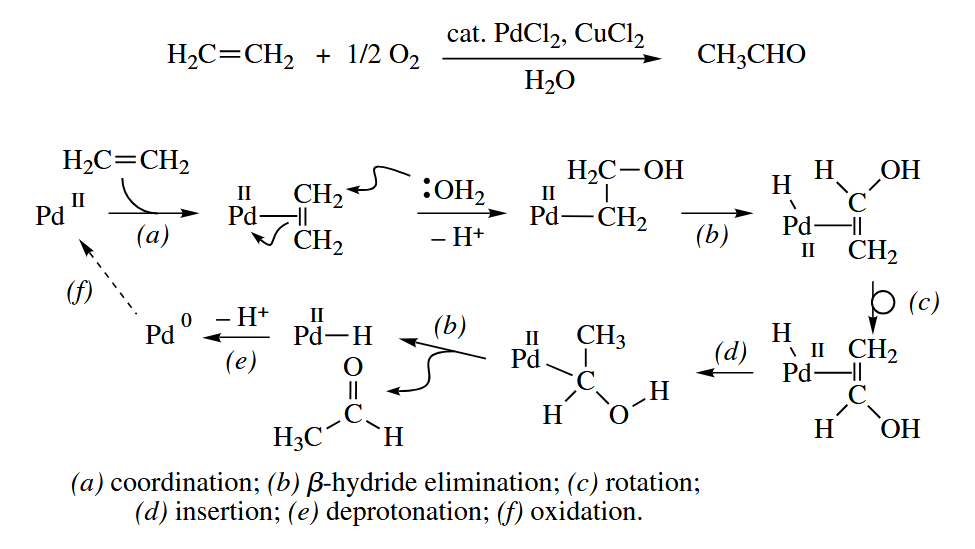
For the reaction to be catalytic in Pd, the Pd(0) has to be reoxidized back to Pd(II). Two equivalents of convert Pd(0) to Pd(II) and produce 2 CuCl. The Cu(I) is then reoxidized back to Cu(II) by .
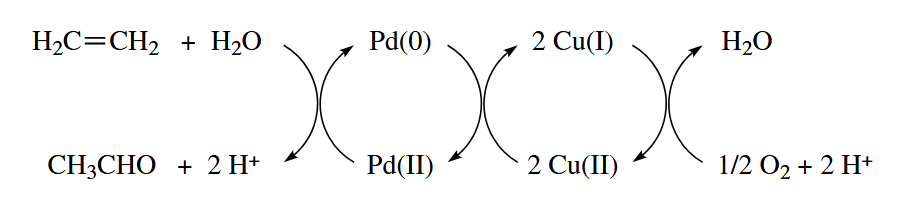
6.3.7 Tebbe Reaction (Ti)
The Tebbe reaction is an early-metal organometallic version of the Wittig reaction. A carbonyl compound is converted to the corresponding methylene compound by treatment with the Tebbe regent, which is a complex of with .
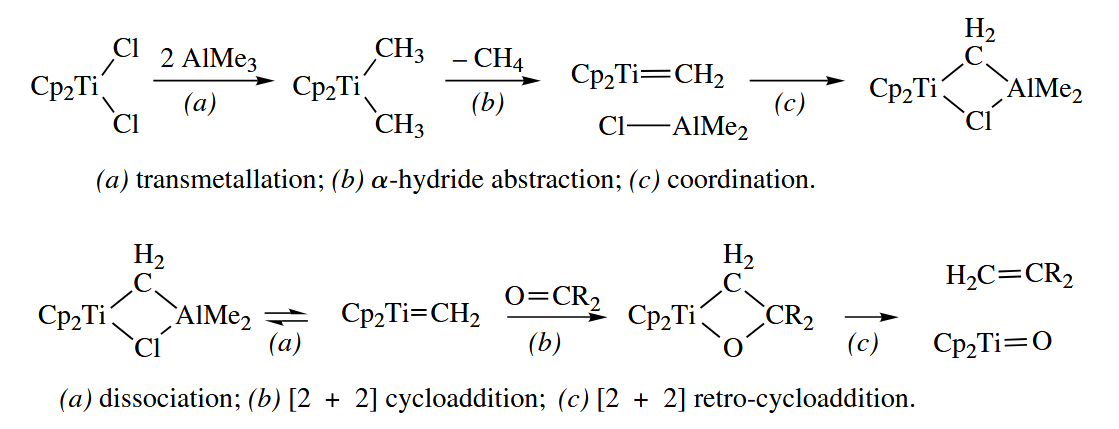
Unlike the conventional Wittig reaction, the Tebbe reaction works well with esters. However, Tebbe reagents are limited to transferring alkyllidene groups lacking β-hydrogens, such as methylidene and benzylidene.
The Petasis reagent, , is used to carry out Tebbe reactions. It is much easier to prepare and handle than the Tebbe reagent. Also, Petasis reagent bearing alkyl groups that have β-hydrogens, such as , undergo β-hydride abstraction much more readily to give titanacyclopropanes than they undergo α-hydride abstraction.
( Petasis 试剂比 Tebbe 试剂更易制备,且更易操作,而且可以转移具有 β-H 的烷基。有一个小问题是,文中使用 Petasis reaction 来描述这个反应,但是实际上 Petasis reaction 另有所指,是一个多组分反应。通常提到这个类 Wittig 反应说的都是 Petasis reagent。 )
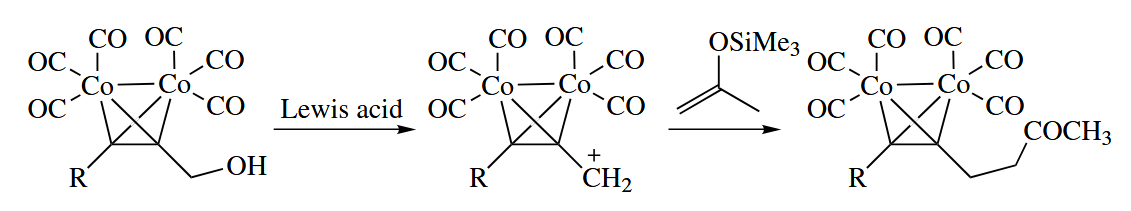
6.3.8 Propargyl Substitution in Cobalt-Alkyne Complexes
Alkyne- complexes have uses in organic chemistry other than the Pauson-Khand reaction. When the complex is formed from a propargl alcohol or ehter, the C-O bond is especially prone to undergo substitution reactions.
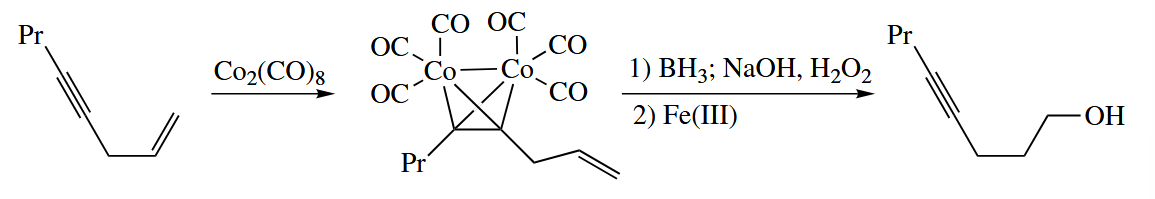
The complex of an alkyl with can also be used to protect an alkyne in the presence of an alkene.
6.4 Rearrangement Reactions
6.4.1 Alkene Isomerization (Rh)
Wilkinson’s catalyst catalyzes the isomerization of an alkene to its thermodynamically most stable isomer. The Wilkinson’s catalyst, though, lacks a Rh-H bond into which an alkene can insert. The reaction may proceed by oxidative addition to an allylic C-H bond, then reductive elimination at the other end of the allylic system.
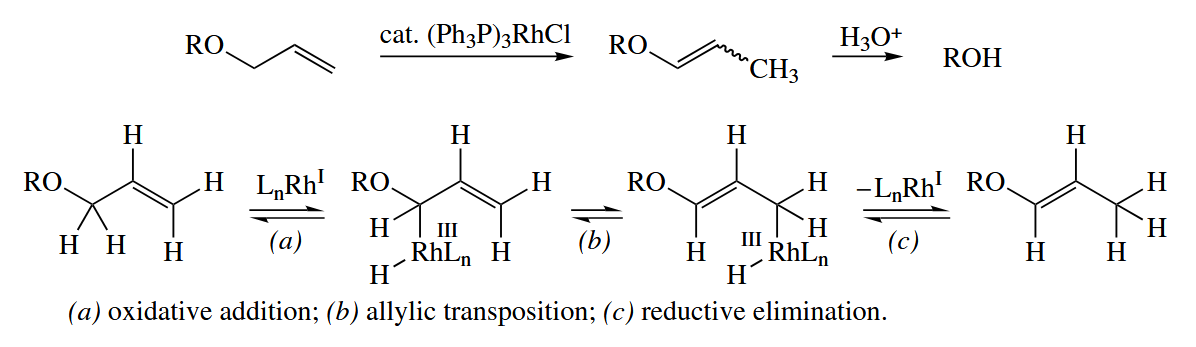
6.4.2 Olefin and Alkyne Metathesis (Ru, W, Mo, Ti)
In the olefin metathesis reaction, the alkylidene fragments of two alkenes are swapped to give two new alkenes.
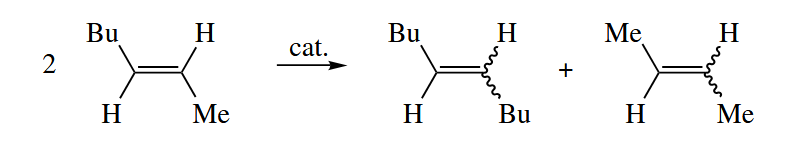
This remarkable reaction is catalyzed by early and middle metals, primarily complexes of Ti, Mo, W, and Ru. All olefin metathesis catalysts either have a M=C π bond or are converted into compounds that have a M=C π bond under the reaction conditions.
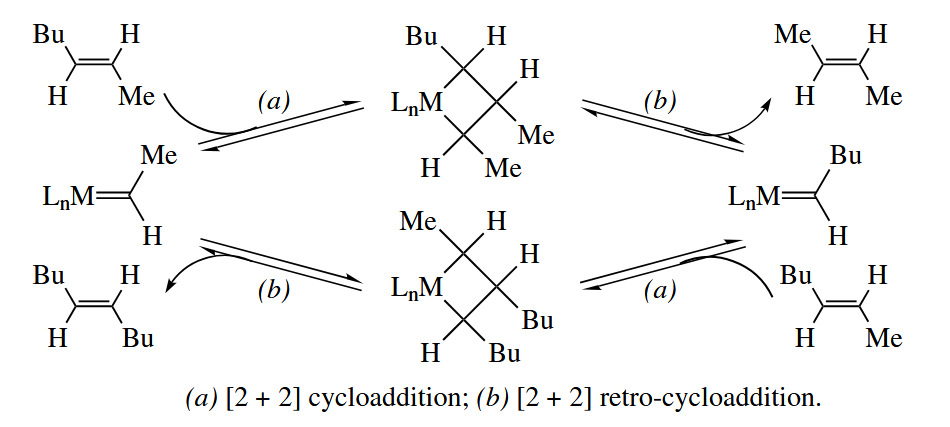
One of the most widely used variations of olefin metathesis is the ring-closing metathesis reaction (RCM).
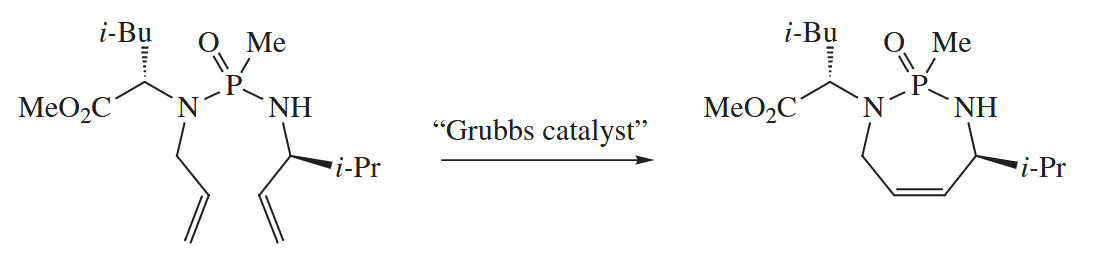
Even more recently, alkyne metathesis catalysts have been developed and applied to organic synthesis. The mechanism of alkyne metathesis also consists of a series of [ 2 + 2 ] and [ 2 + 2 ] retro-cycloadditions.
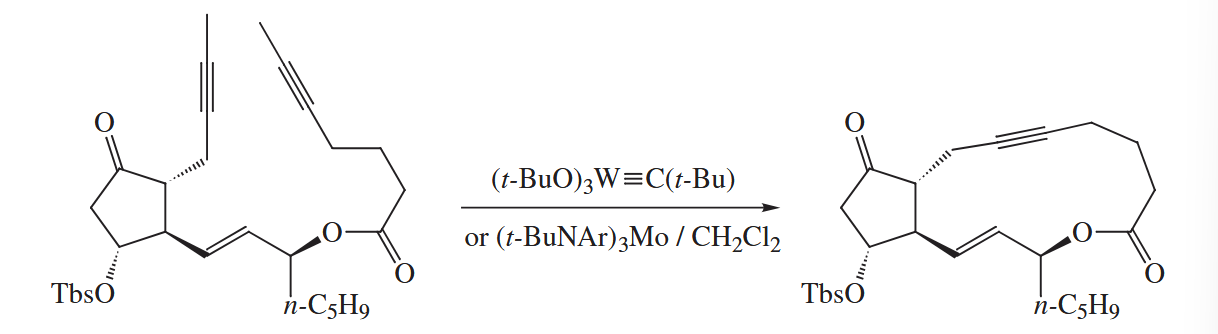
6.5 Elimination
6.5.1 Oxidation of Alcohols (Cr, Ru)
Cr(VI) reagents are widely used to oxidize alcohols to aldehydes/ketones or carboxylic acids. Commonly used reagents include , Jones reagent, PDC, and PCC.
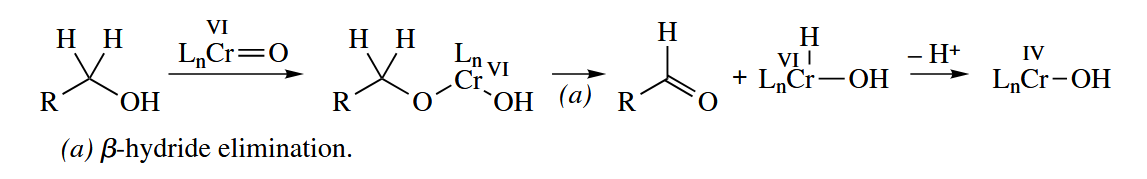
Practical Cr-catalyzed oxidations of alcohols have not been adopted widely, but TPAP () catalyzes the oxidation of alcohols to aldehydes by a stoichiometric oxidant such as NMO, , or itself.
6.5.2 Decarbonylation of Aldehydes (Rh)
Wilkinson’s catalyst mediates the decarbonylation of aldehydes.
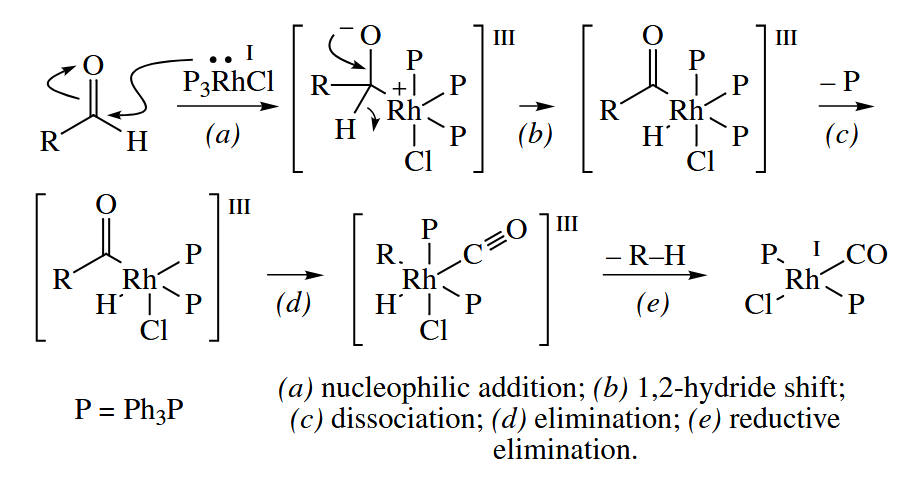
The reaction is stoichiometric (not catalytic) in Rh, as the product complex is inert toward oxidative addition. The electron-withdrawing CO group makes the Rh complex too electron-poor.
6.4 Summary
Metals catalyze a diverse set of reactions, and it is difficult to make generalizations, but some basic principles can be kept in mind.
- Any reaction involving a metal should be one of the typical reactions that have been discussed. 有金属参与的反应应当可归类为前文讨论的经典反应中的一种。
- Ligand association, dissociation, and substitution processes are facile, so the exact number of ligands on a metal center is usually not a major concern when mechanisms and catalytic cycles involving transition metals are drawn. 配体的配合,解离以及取代反应都很容易发生,因此金属中心的配体数在写反应机理时通常不太重要。
- The key step of reactions that involve addition of X-Y across a π bond is insertion of the π bond into an M-X bond. The M-X bond is often generated by oxidative addition of M across an X-Y bond. X-Y 加成到 π 键上的关键步骤在于 π 键对 M-X 键的插入,而 M-X 又是从 X-Y 氧化加成到金属生成的。(氧化加成→配位→插入→还原消除)
- Substitution reactions of organic halides proceed by oxidative addition to the C-X bond. 有机卤化物的取代反应是通过 C-X 键对金属的氧化加成实现的。
- Metals are especially prone to undergo σ-bond metathesis reactions. 金属尤其容易发生 σ 键交换反应。
- Compounds with M=X bonds tend to undergo [ 2 + 2 ] cycloadditons, and metallacyclobutanes tend to undergo [ 2 + 2 ] retro-cycloadditions. 含 M=X 双键的化合物倾向于发生 [ 2 + 2 ] 环加成,而金属杂环丁烷倾向于发生逆 [ 2 + 2 ] 环加成。
- Third-row transition metals tend to undergo α-insertion and α-elimination reactions. 第三行(即第六周期)过渡金属倾向于发生 α-插入和 α-消除。
- Palladium is used in a wide variety of popular reactions with diverse mechanistic pathways. How do you know how to start when facing a Pd-catalyzed reaction? 金属钯被用于多种常见的反应,往往会有多种多样的机理,对于一个钯催化反应,你知道从哪里开始入手吗?
When a leaving group (usually attached to or , allylic, or benzylic) undergoes substitution by a nucleophiles, the first step of the catalytic cycle is usually oxidative addition of Pd(0) to the C-X bond. (In hydrogenolysis, though, the oxidative addition step is preceeded in the catalytic cycle by generation of the intermediate.) 当一个离去基团(通常是连在烯基、炔基、烯丙基或苄基上)被亲核试剂取代,催化循环的第一步通常是对 Pd(0) 的氧化加成。(典型例子如 Suzuki 反应;另外催化氢化稍有不同。)
When nonpolar X-Y is added across a π bond, the first step of the catalytic cycle is usually oxidative addition of Pd(0) across the X-Y σ-bond. However, when a nucleophile adds to a π bond, a Pd(II) complex of the π bond may be the first intermediate. 当非极性的 X-Y 加成到 π 键上,第一步通常是对 Pd(0) 的氧化加成。但是当一个亲核试剂加成到 π 键上,第一个中间体可能是 π 键对 Pd(II) 配位的复合物。(前者很常见了,典型反应如 Stille 偶联等;后者可以参考 6.3.6 节,烯烃本身是富电子的,首先就要配位得到一个烯烃-金属复合物让烯烃缺电子,因此才易于受到亲核试剂的亲核进攻。)
The catalytic cycles of all Pd-catalyzed reactions other than nucleophilic substitutions at alkenes begin with Pd(0), even when a Pd(II) complex is added to the reaction mixture. 除了烯烃的亲核取代外的所有 Pd 催化的反应都是从 Pd(0) 开始,即使你向反应混合物中加入 Pd(II) 也会被还原后再开始催化循环。(Pd 催化的亲核取代反应见 6.3.6 节。)
本章对金属有机反应做了一个简单的介绍,再次强调本章重点在于“写出给定反应的机理”,而不是“预测反应进行”或“解释反应的选择性”,因此许多内容是浅尝辄止或者缺失,需要靠其他专门讲金属有机的教材补充。
金属有机和基础有机有着很多相似,但也有相当大的不同,在金属有机的反应机理中不太强调电子流动,而是注重于用各种基本反应构建出催化循环,因此在机理的训练中需要能够对基本反应烂熟于心,同时熟悉常见反应的催化模式。因为很多反应虽然底物不同,金属不同,但是内在的催化模式却是一样的,掌握了模型后有助于举一反三。
Chapter VI
1.Palladium complexes are versatile and popular catalysts, catalyzing a wide variety of transformations in good yield under mild conditions. Draw mechanisms for the following Pd-catalyzed reactions.
_270bd8f6715d9.png)
Ans
_8e534f54f1c5f.png)
烯丙基位的离去基团在 Pd(0) 配合物的作用下易于发生异构化消旋(epimerization)。首先是 Pd(0) 对烯丙基氮杂环丙烷的氧化加成,其通过类 SN2 的方式从背面取代,并随之形成烯丙基 Pd 复合物,然后 C-C 键旋转,N 原子与 Pd 配位,再还原消除得到异构化产物。常规的烯丙基 Pd 配合物会经历两次 SN2 反应实现构型保持,本题中则有所不同。
_756bbf789a59e.png)
Ans
_80fb09d7c61d3.png)
注意到 C5 连接了两个吸电子基,因此 C5 的 H 酸性非常强,脱去质子后可以作为亲核试剂,但是另一个分子的亲电性不足,因此正好可以与 Pd 配位实现这一要求。注意 Pd(0) 首先作为碱夺去酸性很强的氢,然后 Pd(II) 物种活化炔烃。
_9cb82dbb2b868.png)
Ans
_06866133de051.png)
这个反应在表观上看起来像一步烯丙位羰基插入 + 一步形式上的 Pauson-Khand 反应,在具体写机理时从烯丙基取代入手似乎更合理一些,因为 Pd(0) 的取代需要烯丙基活化,随后就是按部就班把环关上,把羰基插进去。注意该反应副产物为乙酸,因此酸性条件催化的酯化机理更合适。
_13e66a15668d0.png)
Ans
_ed2c073348773.png)
依然是炔丙位离去基团被取代,随后发生羰基插入,再接着发生 Suzuki 偶联,最后通过电环化关环(或者分步的 Michael 加成)得到产物。
_6fb480760e2e2.png)
Ans
_b2bc0b179142b.png)
本题与 1-(g) 题类似,都有一个酸性很强的 H,不过本题反应体系中有 OAc 可以作为碱攫氢,Pd(II) 盐则活化双烯使得其易于被负离子进攻。另一个立体化学的细节是,Pd(II) 始终在六元环面的一侧,因此处在六元环对位的 OAc 和 烷基构型上也呈顺式。
2. Many transition metals other than Pd also catalyze organic reactions. Draw mechanisms for the following meetal-catalyzed reactions.
_9d35552a08d84.png)
Ans
_6e8bacdf237a0.png)
本题的难点在于意识到 Rh(I) 能够对醛基氢做氧化加成,后面的步骤就很自然了。这种性质也使得 Rh(I) 能使羰基化合物脱羰基,但是脱羰反应并不是催化反应,而是需要消耗当量 Rh(I) 催化剂。
_8c420dd50e9c1.png)
Ans
_277277d2175ea.png)
本题类似于 Jacobsen 环氧化,基本模式是形成 M=X 双键,随后 [2+2] 再还原消除得到杂原子三元环(也有可能是自由基机理)。本题答案与正文 6.6 题答案类似,注意 Cu 的价态应当是在 Cu(I) - Cu(III)。
_84d6e6f22059e.png)
Ans
_1a92561c96189.png)
反应通过两步 Y 复合物的 σ-metathesis 完成关环和硅基化,Y 作为典型的 early metal,在成键时有明显的离子键特征,因此反应的驱动力可能来自于从 Y-C 键形成更强的 Y-H 键。
3. Metal-mediated reactions are not as popular as metal-catalyzed ones, but they are still indispensable for some transformations. Draw mechanisms for the following metal-mediated reactions.
_023fb4eecf691.png)
Ans
_5be054873c18c.png)
六羰基二钴可以和炔烃结合,可以作为炔烃的一种保护基,在本题中则能够减小炔基的张力对形成八元环的限制。
_3908402cbf8e6.png)
Ans
_7f79ac471e95a.png)
在完成标号后能够发现本题中共轭二烯烃的两端(C1 和 C4)均发生了亲核类型的反应,这与常规的二烯烃反应性(比如 1,4-加成)不同,所以 Ti 催化剂的作用就是扭转烯烃的反应性,因此选用的是小电负性的 early metal。同时注意 Ti(IV) 需要先通过和格式试剂交换配体才能变成还原剂。
_2ee5dabbf298a.png)
Ans
_fa4682ec0a8a8.png)
本题先用丁基锂对底物做锂卤互换,再和锆试剂交换得到 Zr 复合物,随后发生 β-abstraction 生成锆杂环丙烷结构,然后两个亲核性的碳依次结合两个亲电试剂,此处的反应性类似于双负离子,注意锆杂环丙烷的轨道与苯环 pi 轨道正交,因此只会受到 OMe 的吸电子诱导效应,而没有给电子共轭效应,因此间位的电子密度更大,优先发生对乙腈的亲核加成。
_8840f54f0dc5b.png)
Ans
_8ce06e5a8a548.png)
观察产物结构发现多了一个羰基,那么自然是来自于原料中和 Cr 配位的羰基,通过两步 [2+2] 环加成把与 Cr 相连的片段连接到炔烃上,并且形成五元环,最后羰基迁移插入并发生电环化反应关上六元环。
第六章终于也是写完了,本章作为金属有机的内容尚显基础,更注重于对基元反应的掌握和反应机理的书写,对于更多深入的内容缺乏进一步讨论,因此需要进一步学习才能对现代的金属有机化学反应有一个更好的把握。