《ART》章末总结5
《ART》第五章,隔了有两年没写了,趁这个假期快速结束掉。
5.1 Free Radicals
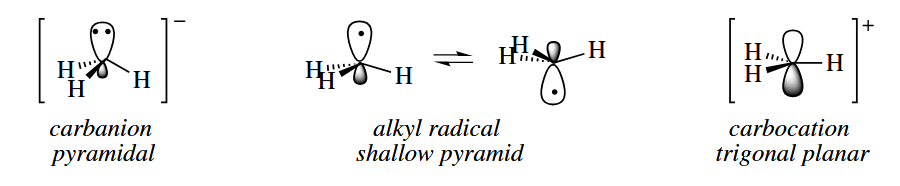
A free radical is a species containing one or more unpaired electrons. Free radicals are electron-deficient species, but they are usually uncharged.
5.1.1 Stability
The alkyl radical is a seven-electron, electron-deficient species. The geometry of the alkyl radical is considered to be a shallow pyramid. The energy required to invert the pyramid is very small, in practice, one can usually think of alkyl radicals as if they were -hybridized.
Both alkyl radicals and carbocations are electron-deficient species, and the structure features that stabilize carboncations also stabilize radicals. However, there are two major differences between the energy trends in carbocations and alkyl radicals.
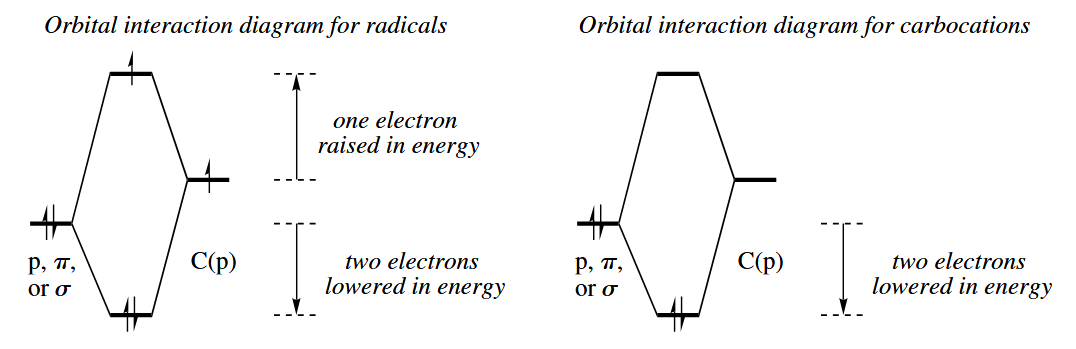
Alkyl radicals are generally not as high in energy as the corresponding carboncations. But the amount of extra stabilization that adjacent groups provide to radicals is not as great as that which they provide to carbocations.
Neutral free radicals are electron-deficient, so radicals centered on less electronegative elements are lower in energy than radicals centered on more electronegative elements. The order of stability for halogens it is I>Br>Cl>F.
Unlike carbocations, free radicals are stabilized both by electron-rich π bonds, and by electron-poor π bonds. An electron-rich π bond is more stabilizing than one that is electron-poor.
When a radical is substituted by both an electron-donating and an electron-withdrawing group, the
total stabilization is greater than the sum of the parts in a phenomenon called the captodative effect.
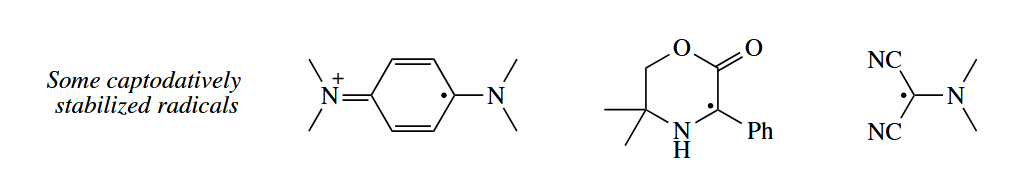
( captodative 效应描述的是自由基旁边的吸电子基团和给电子基团的协同稳定效应,该名字的来源是吸电子基团有时会被称为“captor”,而给电子基团则被称作“dative”,同时连有吸电子基和给电子基的烯烃有时会被称为“captodative”。 )
Tables of homolytic bond strengths (bond dissociation energies, BDEs) offer a good guide to the relative energies of radicals.
Free radicals can also be stabilized kinetically by steric shielding of the radical centers. Such free radicals are called persistent. Examples of persistent free radicals include perchlorotrityl, galvinoxyl, and the radical derived from BHT, an antioxidant that is used as a food preservative.
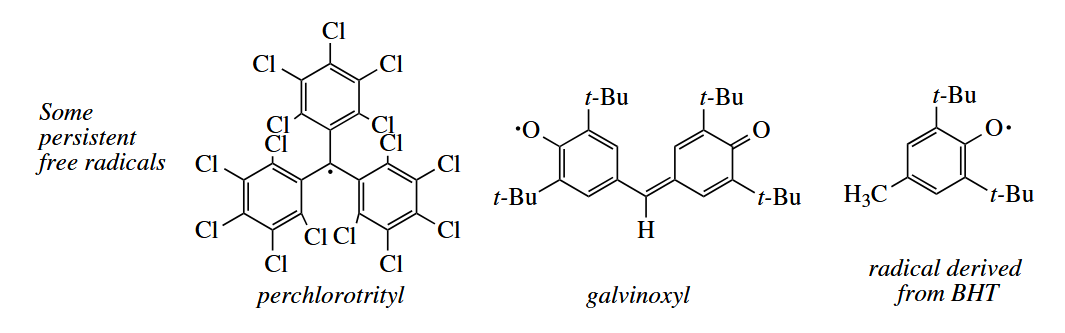
( The persistent radical effect(PRE)效应往往用于解释自由基的选择性交叉偶联,这种动力学稳定的自由基物种在发生自由基反应时不会自身耦合,而总是与其他自由基物种发生偶联。 )
5.1.2 Generation from Closed-Shell Species
Free radicals can be generated from closed-shell species in four ways: σ-bond homolysis, photochemical excitation of a π bond, one-electron reduction or oxidation, and cycloaromatization.
- Sigma-bond homolysis is a very common way to generate free radicals. The σ bond is usually a heteroatom–heteroatom bond such as N–O or Br–Br, but even σ bonds such as C–C or C–N that are normally very strong can homolyze if a very stable fragment is formed or if the bond is very strained.
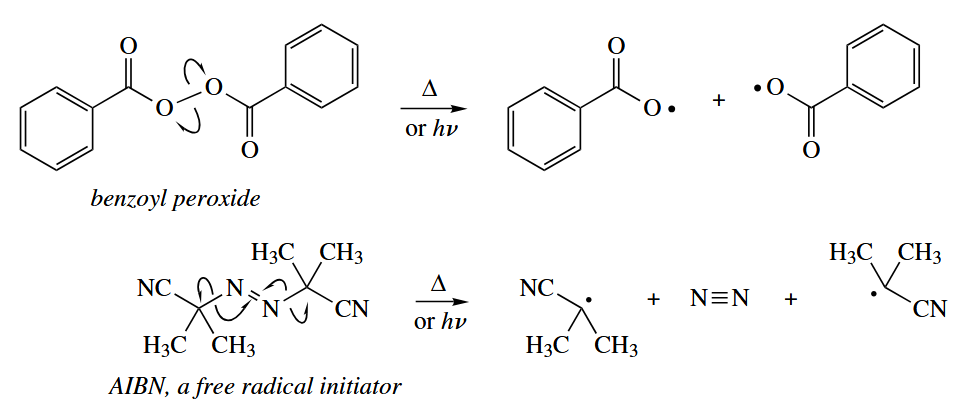
- When light of the appropriate wavelength is allowed to shine on a compound containing a π bond, an electron from the π orbital is promoted to the π* orbital. The product can be considered to be a 1,2-diradical, and it undergoes reactions typical of free radicals.
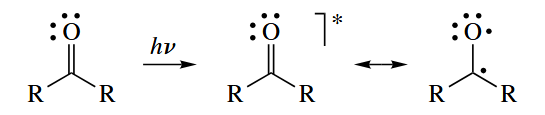
The weaker the π bond, the easier it is to photoexcite.
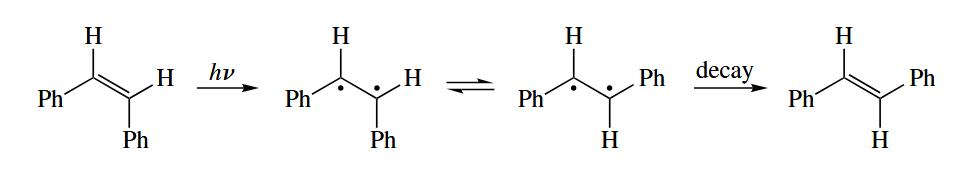
In photoexcited alkenes, there is no π bond can occur. Alkenes can be isomerized from the cis to the trans from photochemically. The equilibrium favors the less sterically encumbered trans isomer.
- A compound with an electron in a high-energy orbital may transfer the electron to a compound that has a lower energy orbital. Most often, the electron donor is a metal or reduced metal salt such as Li, Na, or , but the donor may also be a lone-pair-bearing compound such as an amine or phosphine, especially if it is photoexcited.
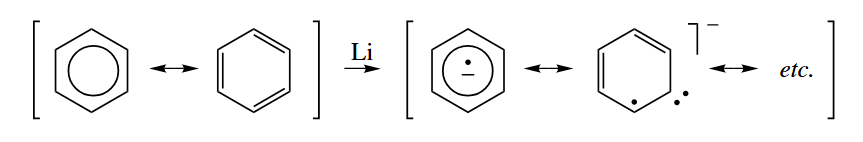
After the electron is accepted, the product is called a radical anion.
One-electron oxidation of organic substrates gives radical cations. Oxidadtion is much less common in synthesis than is reduction. Certain metal salts such as . - The cycloaromatization of certain highly unsaturated organic compounds can give diradicals. The best-known cycloaromatization reaction is the Bergman cyclization of an enediyne.
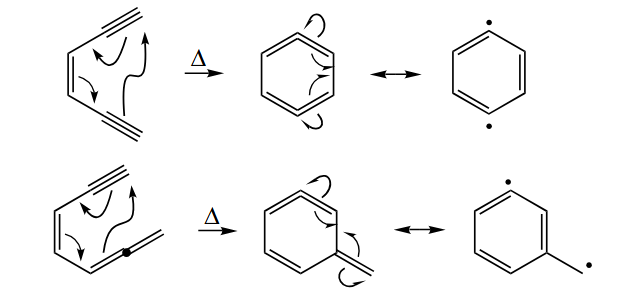
5.1.3 Typical Reactions
Free radicals undergo eight typical reactions: addition to a π bond, fragmentation, atom abstraction (reaction with a σ bond), radical–radical combination, disproportionation, electron transfer, addition of a nucleophile, and loss of a leaving group. The first three are by far the most important.
- A free radical can add to a π bond of a closed-shell species to give a new radical. The radical (X·) addds to a Y=Z π bond to give X-Y-Z·.
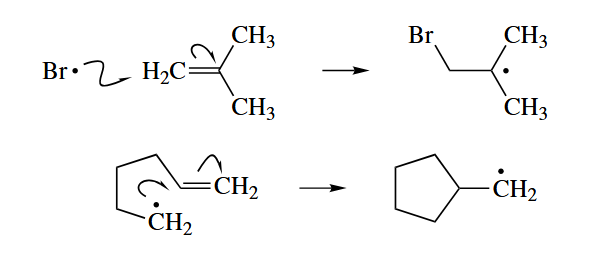
- Fragmentation is the microscopic reverse of addition to a π bond. A σ bond that is adjacent to a radical center homolyzes, and one of the electrons of that σ bond and the former unshared electron together form a new π bond at the former radical center.
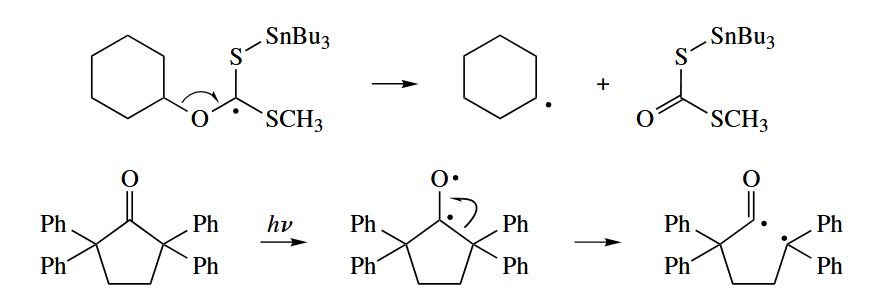
The combination of and is widely used to initiate radical reactions. The mechanism involves two unusual radical reactions.
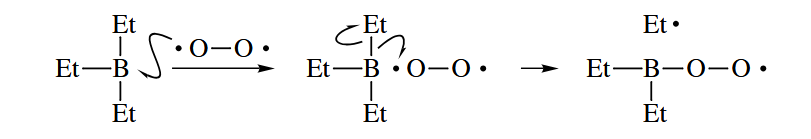
- In an atom abstraction reaction, a radical X· attacks a Y-Z σ bond to give a new closed-shell species X-Y and a new radical Z·.
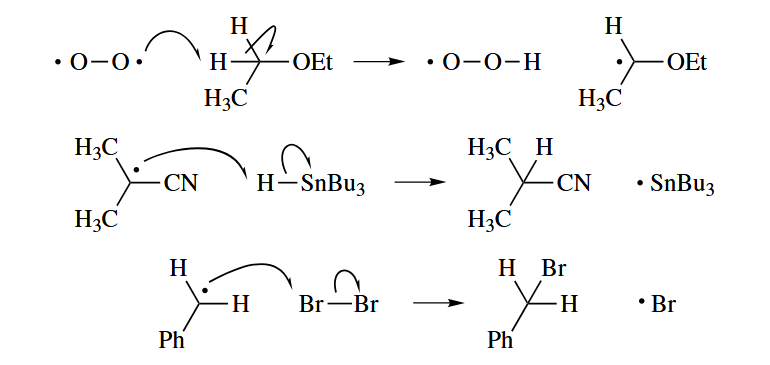
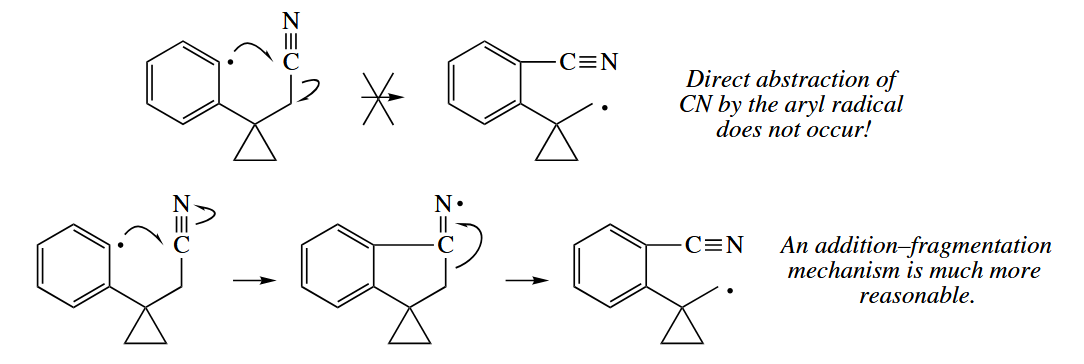
The tributylin radical () is often used to abstract heavy atoms X from C-X bonds. The atom X is usually Br or I, but it may be Se or even S. Abstraction of C does not occur in free-radical reactions. An addition-fragmentation mechanism can be drawn for an apparent atom abstraction of C() or C().
- Two radicals can react with one another in one of two ways: radical-radical combination or disproportionation. The two reacting mode show below.
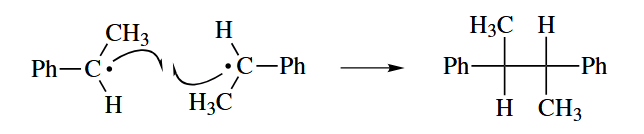
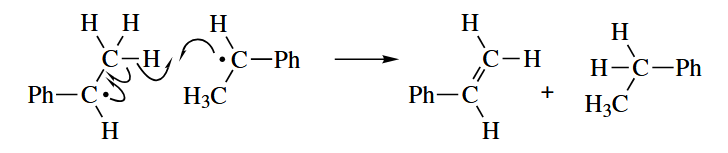
( 在链式机理中,自由基结合和歧化反应通常只有在链终止阶段才会出现,但是有一个例外:氧气参与的自由基反应,因为氧气是双自由基,所以在发生了自由基结合后仍然可以继续发生链转移;而在非链式机理中这两类反应则很常见。 ) - A radical can undergo one-electron transfer (oxidation or reduction) in the presence of an oxidizing or reducing agent to give an even-electron species.
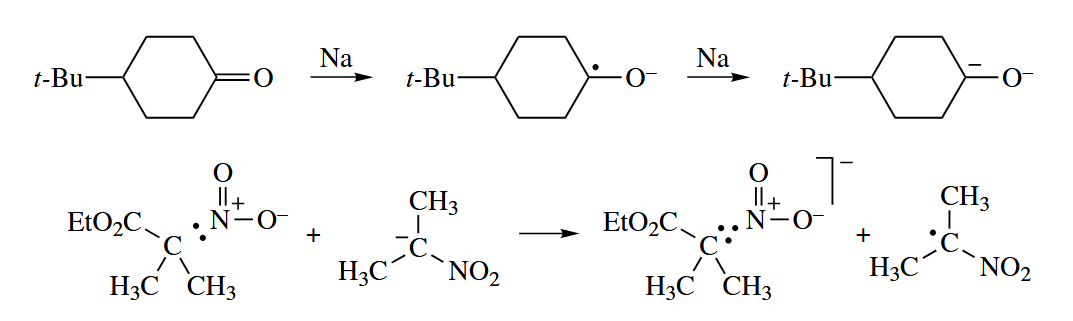
- A radical anion can lose an anionic leaving group to give a neutral free radical, and in the reverse direction, a neutral free radical can combine with an anionic nucleophile to give a new radical anion.
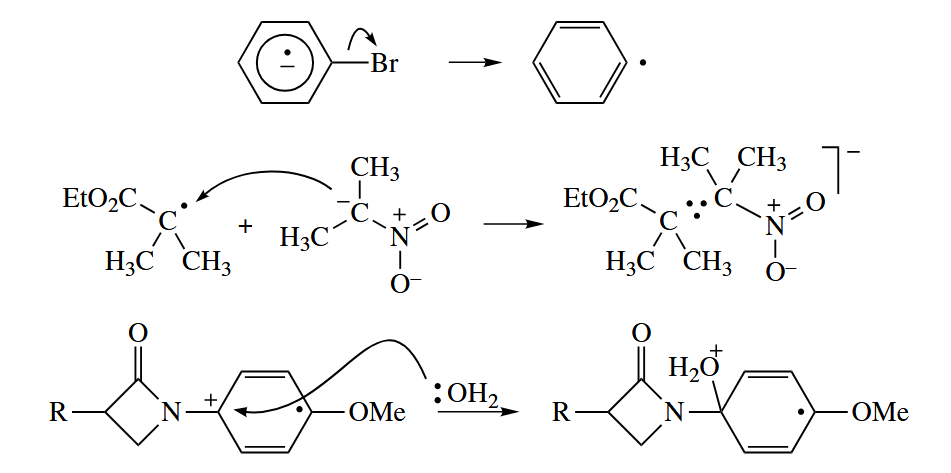
Note that a concerted 1,2-radical shift is allowed only when one of the components can react antarafacially. The 1,2-hydrogen atom shift is geometrically impossible, and the geometric requirements of the 1,2-alkyl radical shift are so stringent that it is not observed.
Unsaturated groups, however, can shift by a two-step addition-fragmentaion mechnism via a discrete cyclopropane radical intermediate.
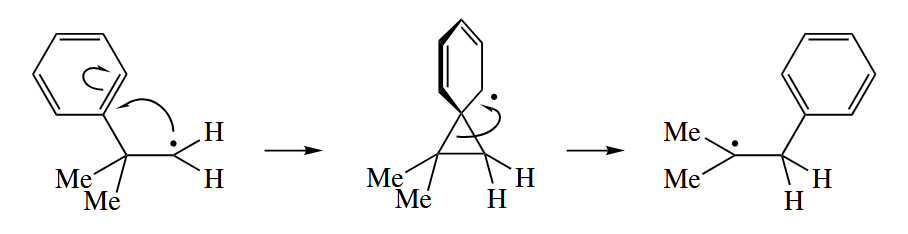
Heavy atoms such as Br and Se can also undergo a radical 1,2-shift by an addition-fragmentation mechanism.
5.1.4 Chain vs. Nonchain Mechanisms
A free-radical reaction may proceed by a chain or a nonchain mechanism. Usually, the reagents or reaction conditions will indicate which type of mechanism is operative.
Reactions involving stoichiometric amounts of one-electron reducing or oxidizing agents such as always proceed by nonchain mechanisms. Reactions that generate two radicals simultaneously and in close proximity, such as cycloaromatizations and many photochemical reactions, also usually proceed by nonchain mechanisms.
All chain reactions require an initiator and there are only a few widely used initiators, so the presence of these initiators is a good sign of a chain mechanism.
Be ware, though, that the (apparent) absence of an initiator does not rule out a chain mechanism. If a free-radical reaction involves a tin compound (e.g. ), it proceeds by a chain mechanism. However, not all reactions involving are free-radical reactions.
5.2 Chain Free-Radical Reactions
5.2.1 Substitution Reactions
Probably the best-known example of a free-radical reaction is the halogenation of alkanes with or NBS. This chain reaction is initiated by homolytic cleavage of induced by light. The propagation part consists of two atom abstraction reactions.
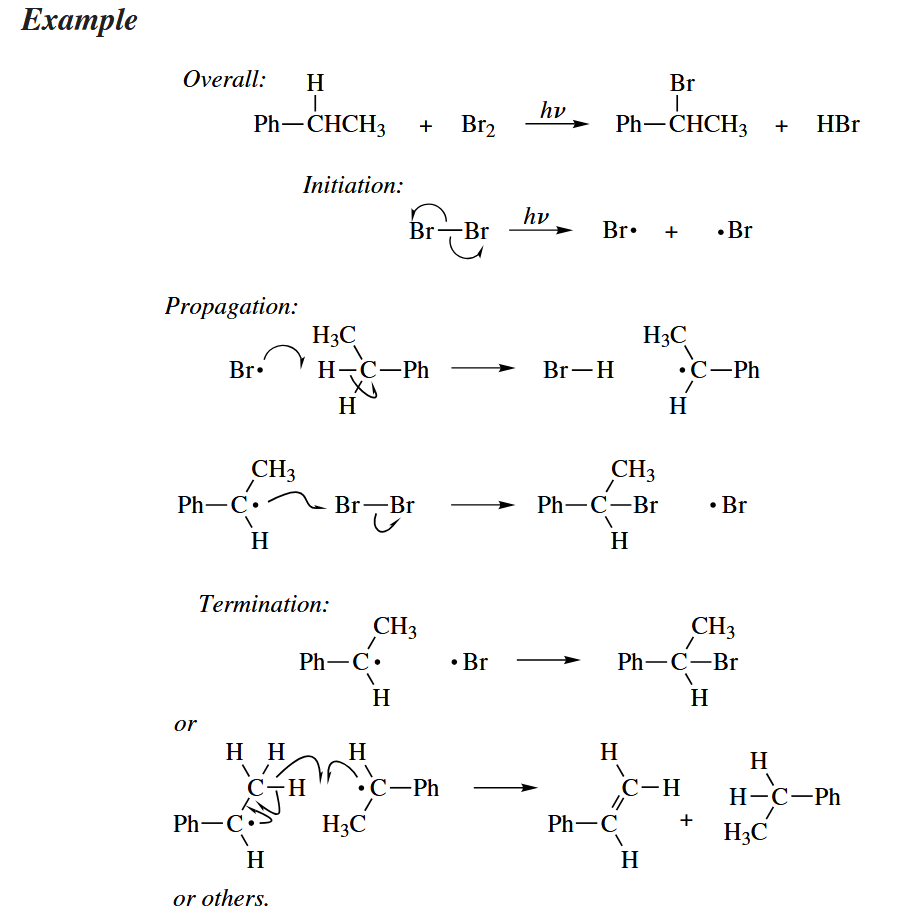
In brominations with NBS, it is thought that is the actual halogenating agent. The is generated by reaction of HBr (the by-product of halogenation) with NBS.
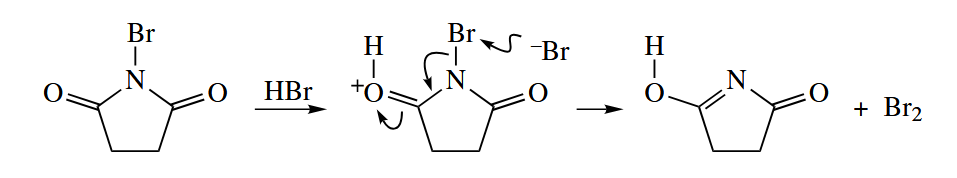
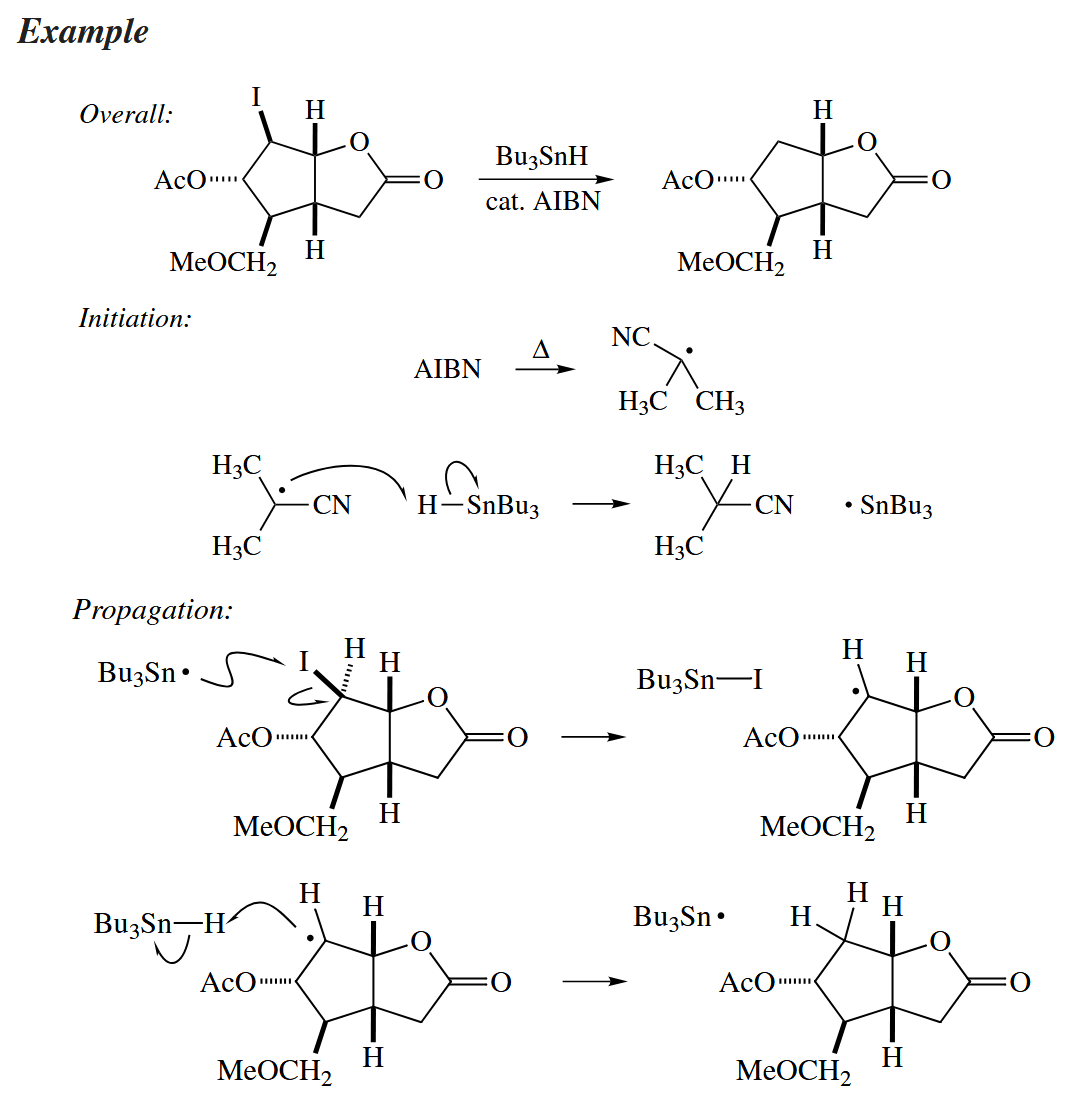
Free-radical dehalogenations are common, too. In these reactions, a C–X bond is replaced with a C–H bond. The reducing agent is most commonly , although or a combination of a catalytic amount of and a stoichiometric amount of may be used instead as environmentally more friendly reagents.
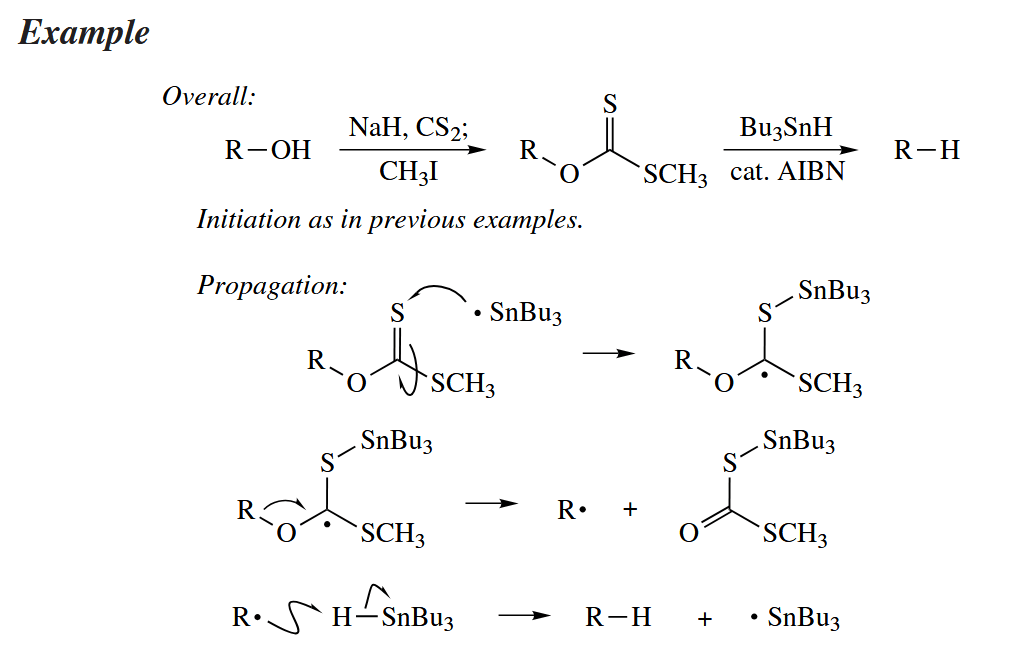
Alcohols are deoxygenated in the Barton-McCombie reaction. The alcohol is first converted into a xanthate or another thiocarbonyl compound, and then the entire functional group is removed by and replaced with H.
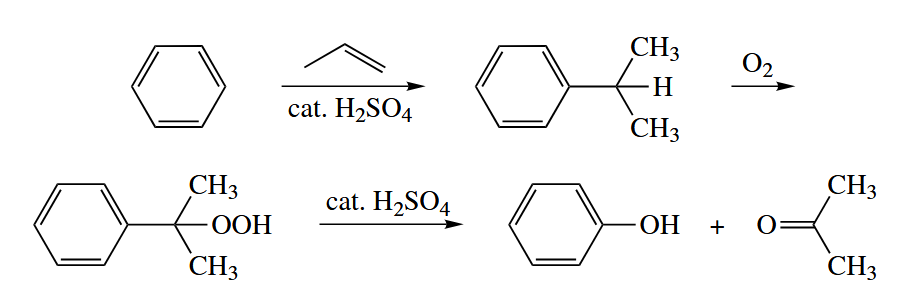
The substitution of H with OOH in alkanes is called autoxidation. Autoxidation proceeds by a free-radical chain mechanism.
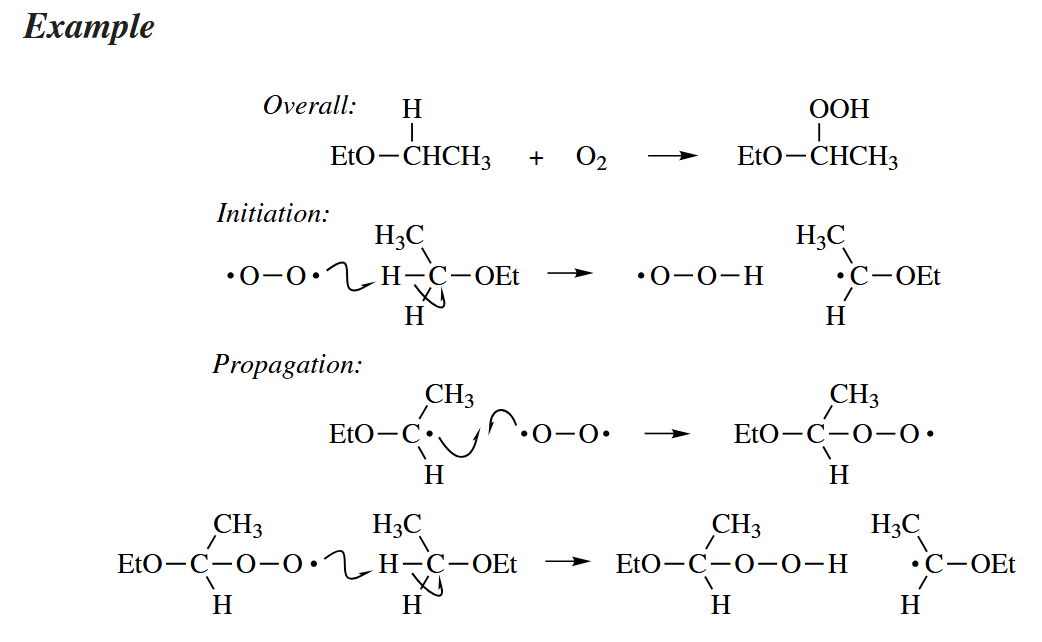
( 值得注意的是,自动氧化过程中存在链式反应中非常少见的自由基结合过程,这一点我们上文也提到了,因为氧气可以作为双自由基参与反应,所以自由基结合不会终止反应。 )
Autoxidation is one of the key steps in the industrial synthesis of phenol and acetone from benzene and propylene.
In the Hofmann–Loeffler–Freytag reaction, an N-chloroammonium ion is converted by a free-radical substitution reaction into a 4-chloroalkylammonium ion, which then undergoes intramolecular substitution to give a pyrrolidine.
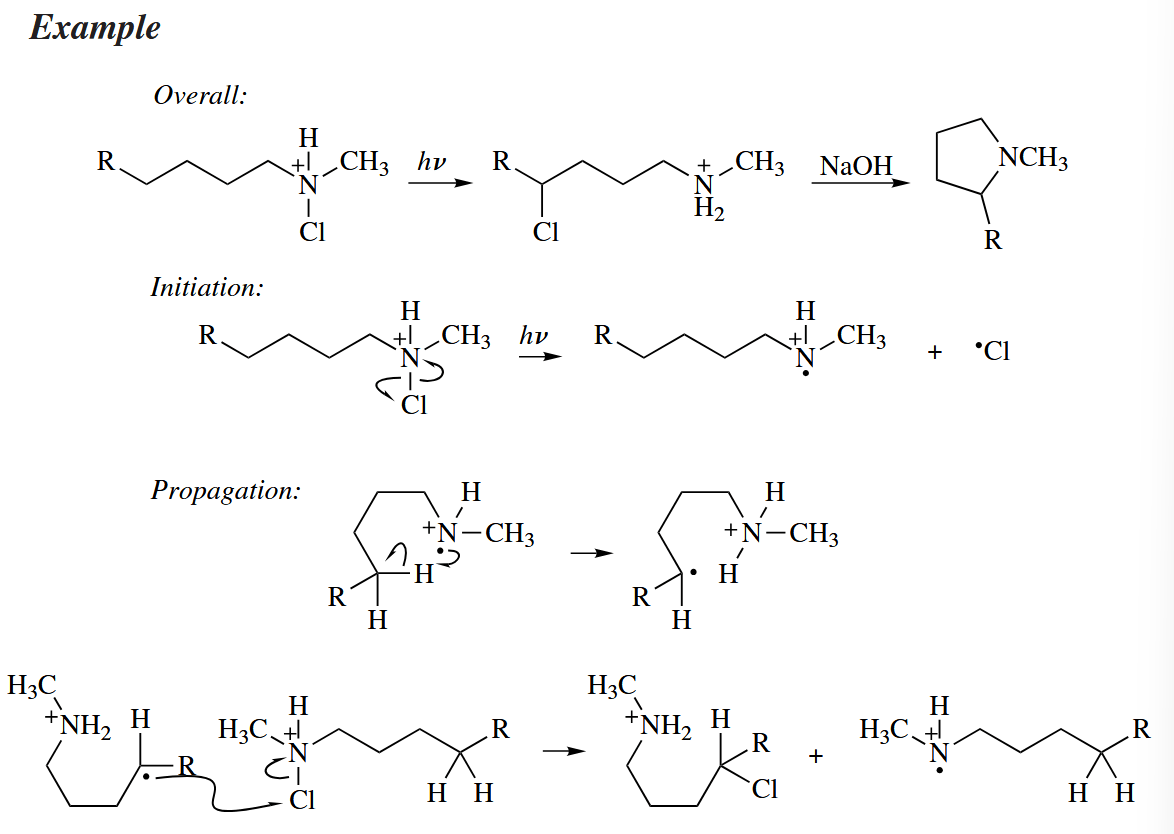
Entropic and stereoelectronic factors make the regioselectivity very high for the C4 hydrogen.
( Hofmnan-Loffler-Freytag 反应可以将烷基 N-卤化物在加热等条件下转化为环胺化合物,反应通常经历一个六元环的椅式过渡态,因而得到五元环产物。在极少见情况下,受化合物骨架刚性的影响,可能得到六元环化合物。 )
5.2.2 Addition and Fragmentation Reactions
The anti-Markovnikov addition of HBr to alkenes was probably the first free-radical addition reaction to be discovered. The anti-Markovnikov regiochemistry derives from the addition of the Br· radical to the less substituted C of the alkene (steric reasons) to give the lower energy, more substituted radical (electronic reasons).
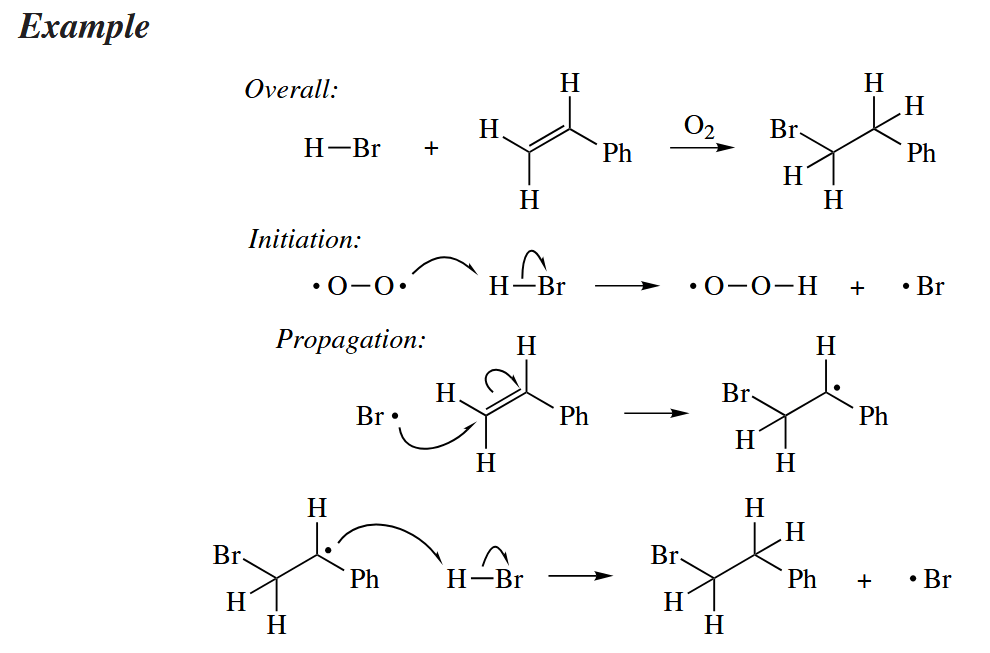
In contrast to HBr, the acids HCl and HI do not undergo free-radical addition to alkenes, even in the presence of peroxides or . Abstraction of H· from HCl is too endothermic, and addition of I· to an alkene is too endothermic.
However, thiols (RSH) add to alkenes by a free-radical mechanism exactly analogous to the addition of HBr. The initiator is usually AIBN or .
Tributylin hydride adds across C=C π bonds by a free-radical mechanism. Addition of across alkynes is one of the best ways of making alkenyltin compounds.
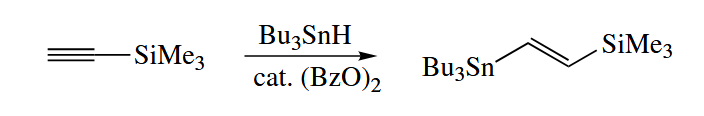
In the laboratory, free-radicl addition reactions often use radicals as key chain carriers. The initiation part of such reactions usually has a radical initiator (often derived from AIBN) abstract H· from to give .
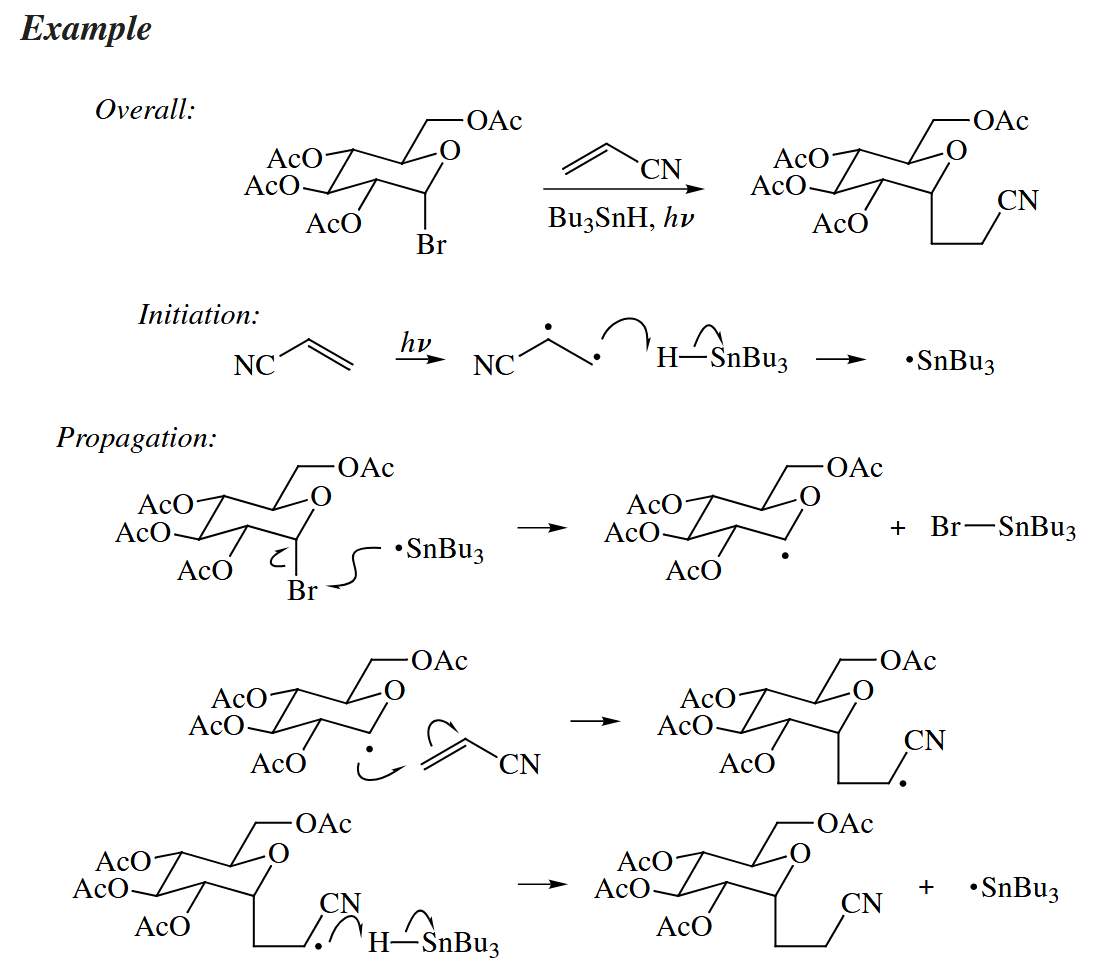
Free-radical cyclization reactions (i.e., the intramolecular addition of an alkyl radical to a C=C π bond) have emerged as one of the most interesting and wide-spread application of free-radical chemistry to organic synthesis.
Free-radical cyclizaitons are useful because they are so fast. In fact, the rate of formation of the cyclopentylmethyl radical is much faster than the rate of cyclization to the lower energy cyclohexyl radical.
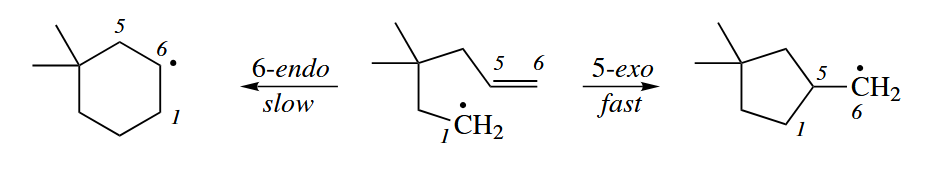
This stereoelectronic effect is derived from the fact that the overlap between the p orbital of the radical and the π* MO of the double bond is much better when C1 attacks C5 than when it attacks C6. The relative rates of 5-exo and 6-endo ring closures are strongly dependent on the nature of the substrate and especially on the amount of substitution on the π bond.
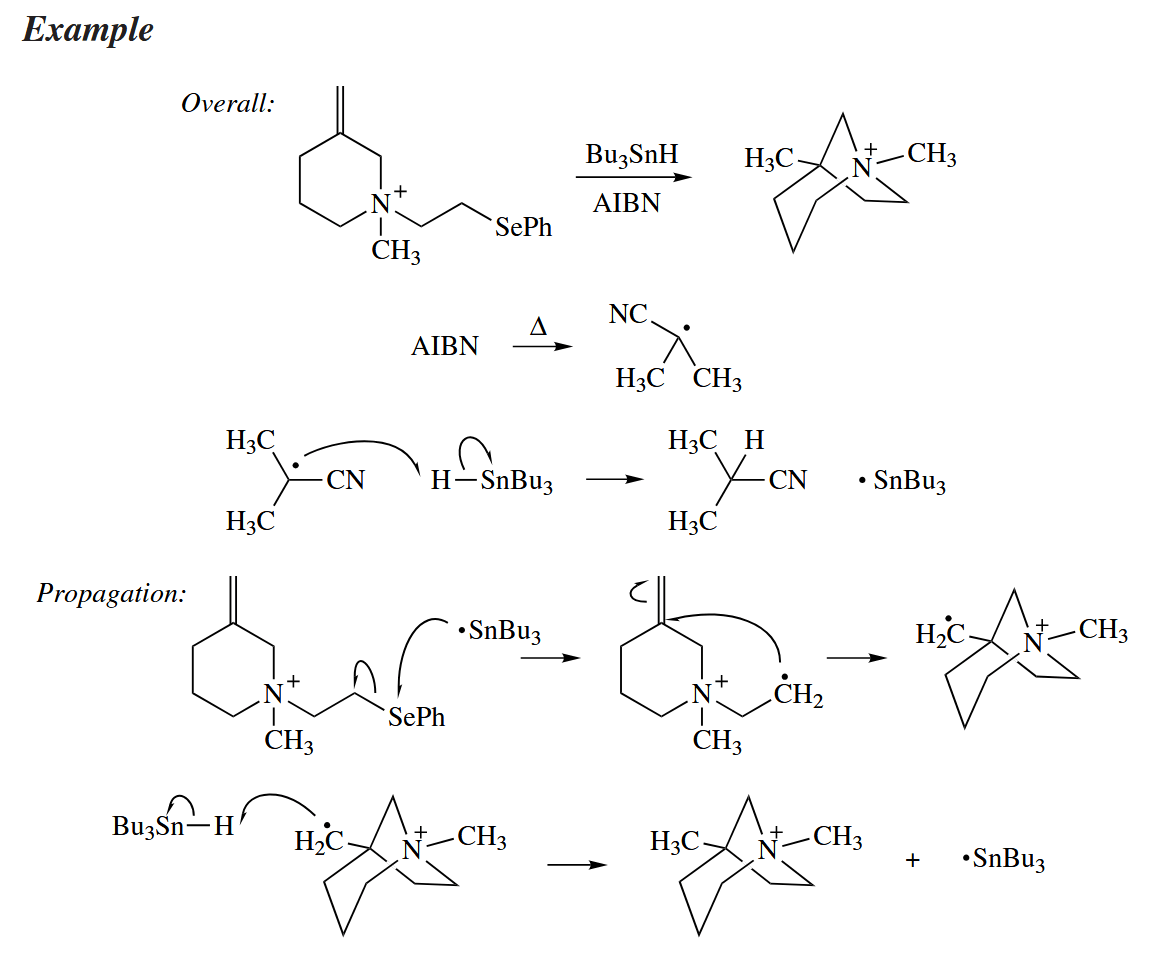
A free radical can add to CO or an isocyanide (RNC) in the course of a free-radical cyclization reaction, too, to give an acyl radical or an iminyl radical.
( 异氰和一氧化碳在参与自由基反应时,可以看作是类卡宾物种,反应生成酰基自由基和亚胺基自由基。 )
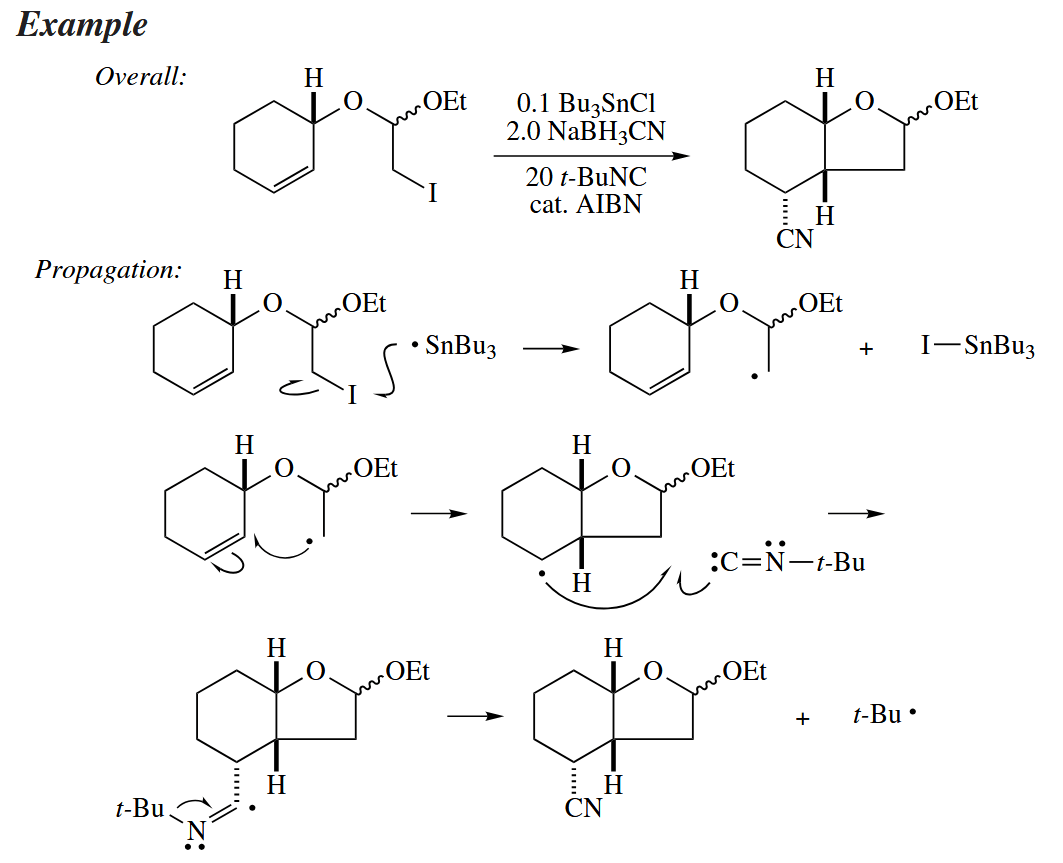
Some free-radical cyclization reactions require only a catalytic amount of or . Such a reaction, instead of replacing a C-X bond with a C-H bond, simply relocates the X atom.
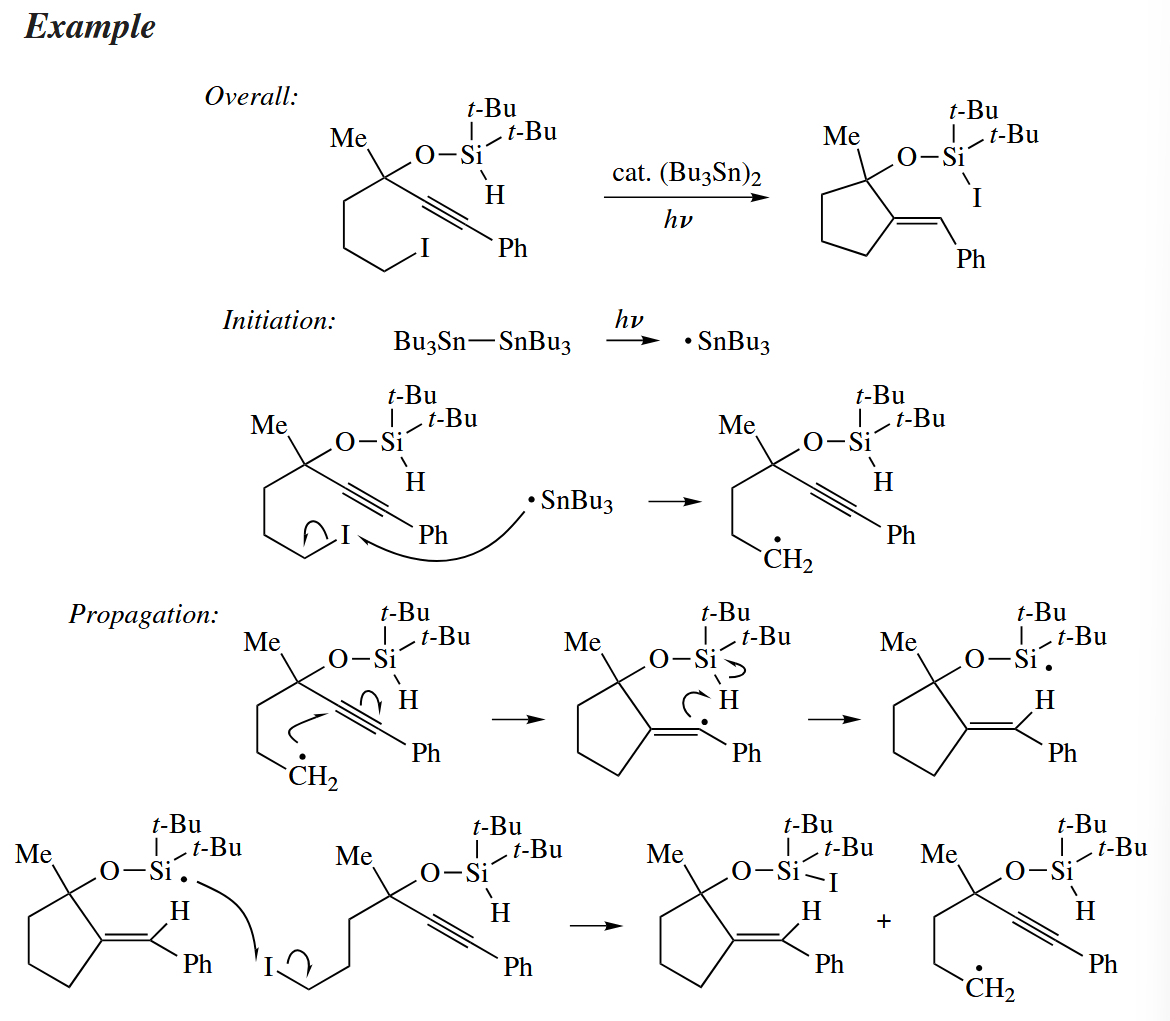
These atom trasnfer cyclization differ from more conventional free-radical cyclizations in that the last step in the propagation involves abstraction of X· from a C-X bond not by but by the cyclized starting material. The last step is kinetically viable only when a stronger bond is made from a weaker one.
Compounds with weak C-H bonds can add to alkenes by a free-radical chain mechanism. Compounds that can add to alkenes in this way include RCHO compounds and 1,3-dicarbonyl compounds.
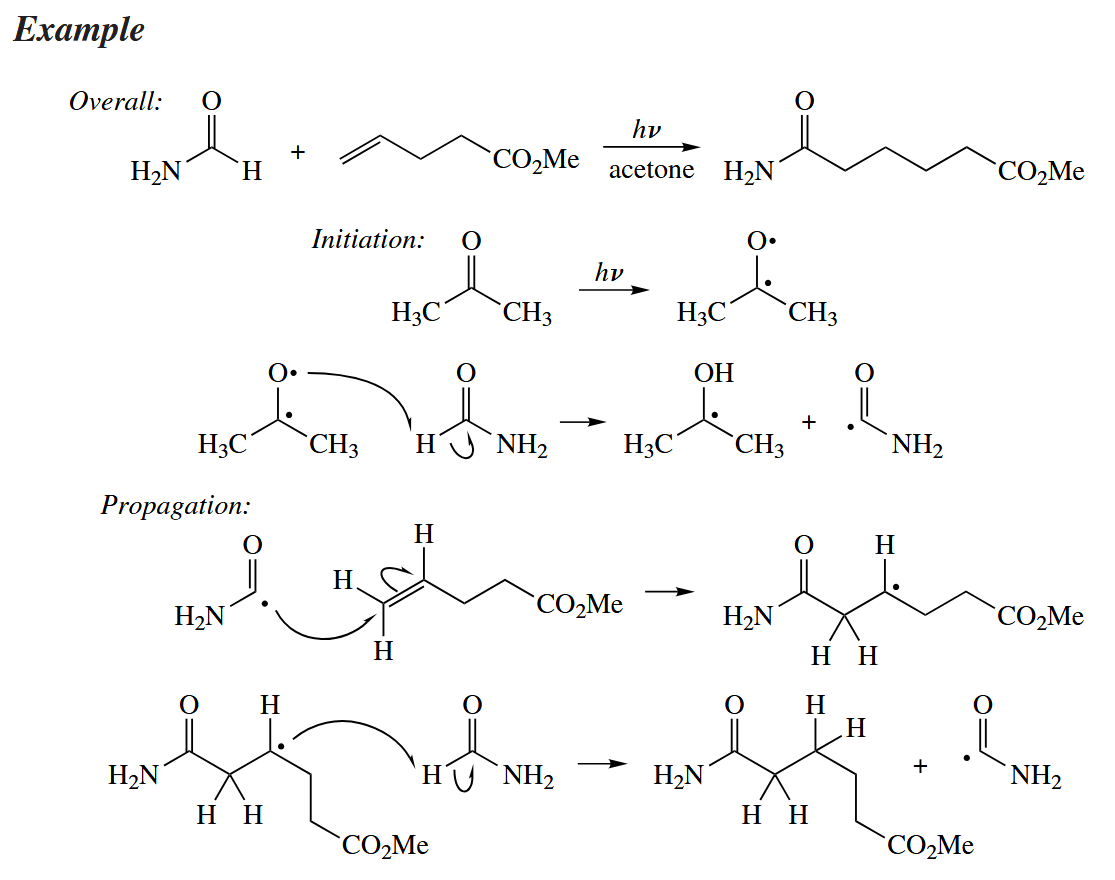
The addition of a free radical to a π bond is sometimes followed directly by fragmentation of the new free radical.
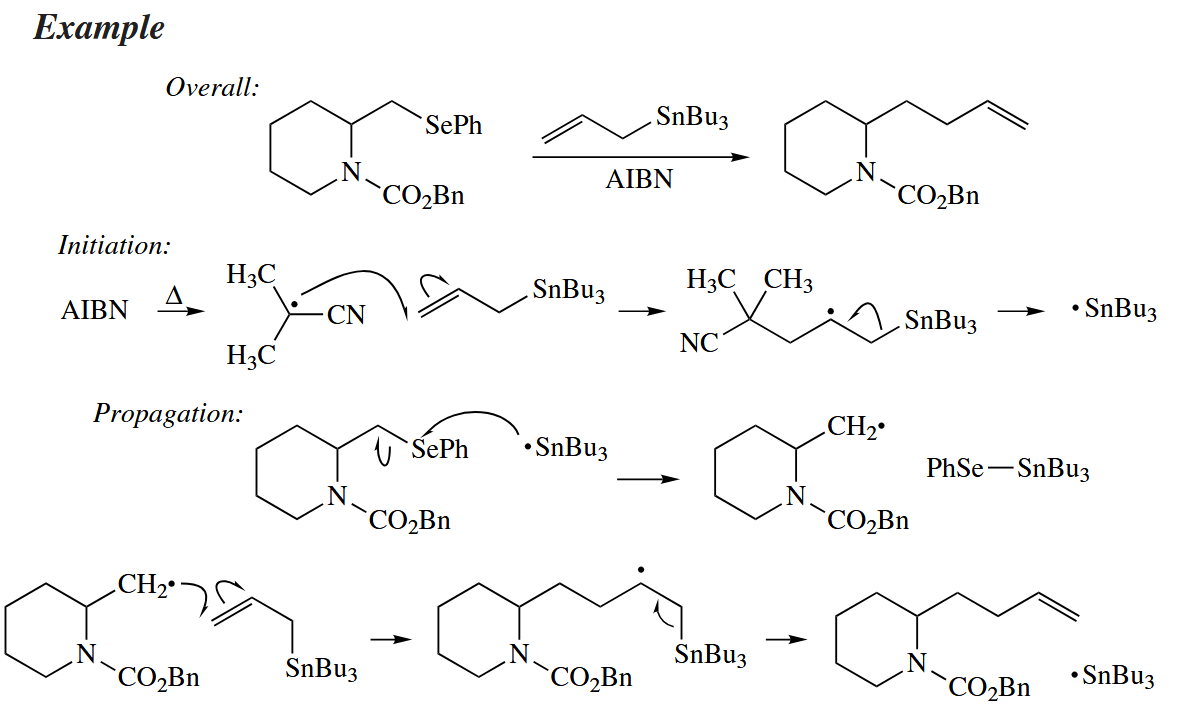
The fragmentation of cyclopropylmethyl radicals to give 3-butenyl radicals is one of the fastest reactions known, occurring at a rate of ! These radical clocks can be used to probe the mechanism of very fast reactions, especially those in enzymes.
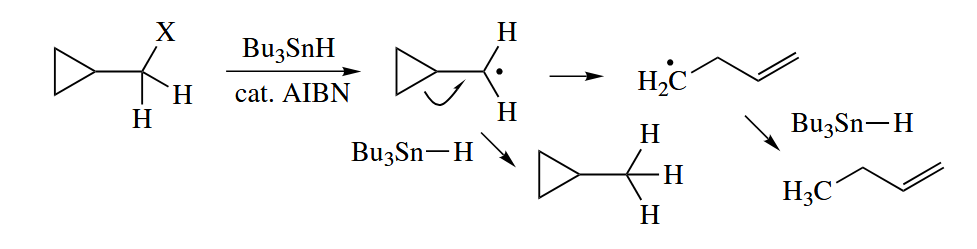
( 自由基钟实验是通过一种已知反应速率的反应,来测定另一个自由基反应的速率。以环丙基甲基自由基的开环反应为例,在自由基中心的旁边连接环丙基,如果反应中间体是自由基,那么可能会产生环丙烷开环物种,根据不同产物的比例就能推得中间体的寿命。 )
In the Hunsdiecker reaction, the silver salt of a carboxylic acid () is treated with to give an alkyl bromide RBr with one fewer C atom. The reaction does not work well with aromatic acids, suggesting that a free-radical mechanism is involved.
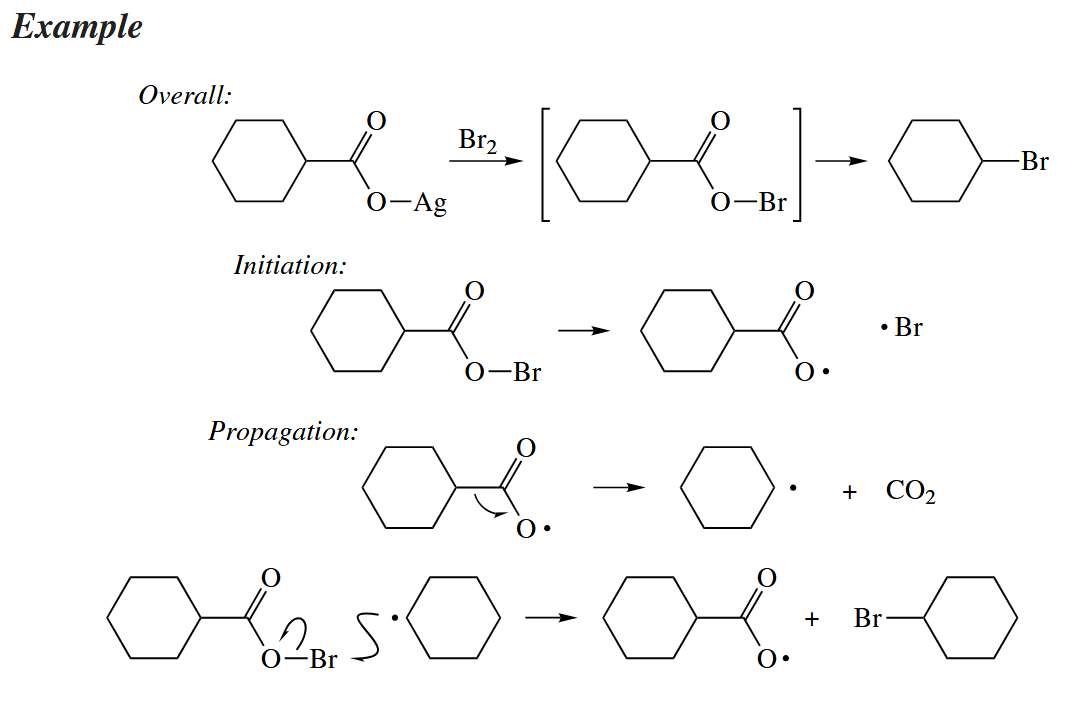
5.3 Nonchain Free-Radical Reactions
The major classes of nonchain free-radical reactions are photochemical reactions, reductions and oxidations with metals, and cycloaromatizations.
5.3.1 Photochemical Reactions
The photoexcitation of a carbonyl compound to give a 1,2-diradical is often followed by a fragmentation reaction. The fragmentation can take one of two courses, called Norrish type I and Norrish type II cleavages.
In Norrish type I cleavage, the bond between the α-carbon and the carbonyl carbon cleaves to give an acyl radical and an alkyl radical.
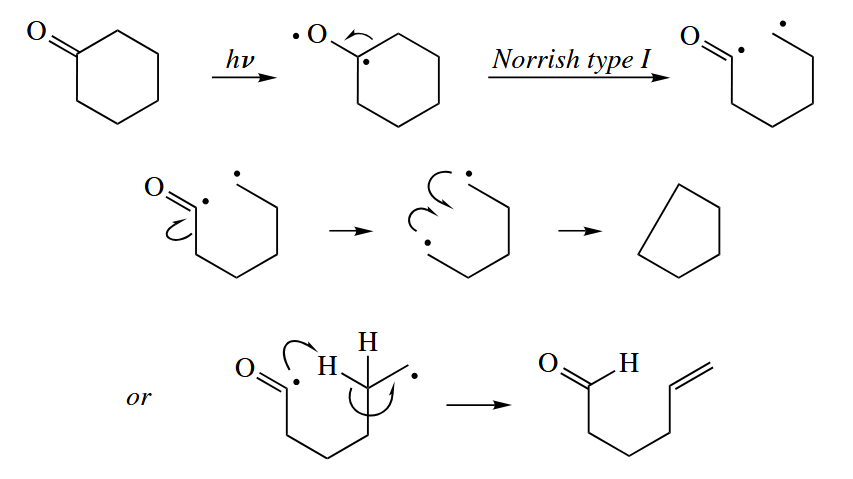
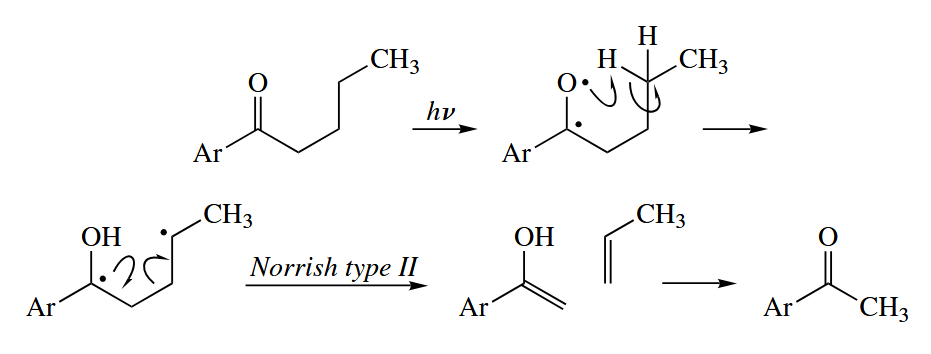
In Norrish type II cleavage, the O radical abstracts H from the γ-carbon in a six-membered TS, and the 1,4-diradical then fragments to give an alkene and an enol, the latter of which tautomerizes to the ketone. The Norrish type II cleavage is closely related to the McLafferty rearrangement that is often seen in the mass spectra of carbonyl compounds.
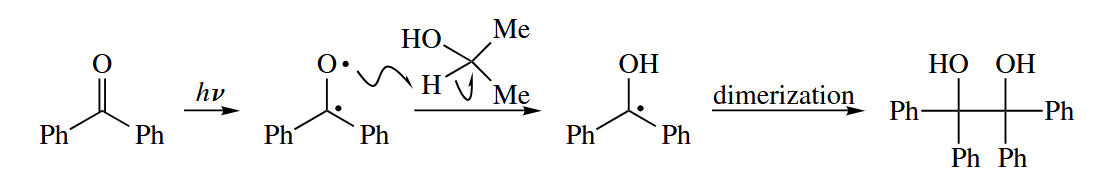
Photoexcitedd ketones can undergo other reactions typical of free radicals. Benzophenone dissolved in isopropyl alcohol undergoes a pinacol coupling merely upon exposure to sunlight.
In Barton reaction, an alkyl nitrite is converted into an alcohol-oxime. The reaction has been used for the remote functionaliztion of hydrocarbons, especially steroids.
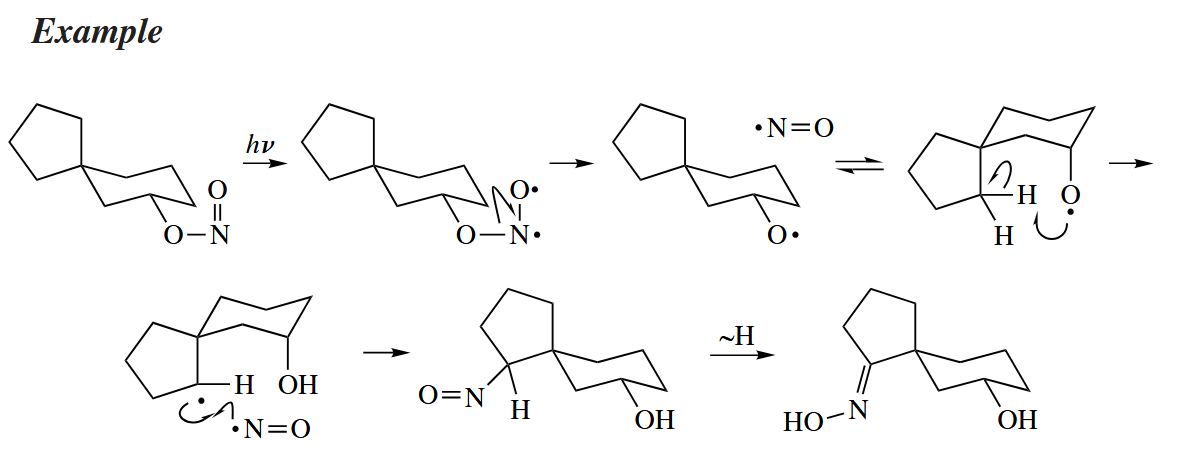
( 该反应和上文提到的 Hofmann–Loeffler–Freytag 反应有异曲同工之妙,关键都在于自由基远程攫氢,后实现官能团化。 )
The photochemically allowed [2+2] cycloaddition reaction of alkenes can be considered to be a radical-mediated process. Spectroscopic measurements at the femtosecond time scale have recently proven that the 1,4-diradical is a true intermediate along the reaction pathway of [2+2] cycloadditions. However, the lifetime of the 1,4-diradical is shorter than the rate of rotation about C–C σ bonds, as [2+2] cycloadditions are stereospecific.
5.3.2 Reductions and Oxidation with Metals
Reductions involving addition of across a π bond follow one or both of the following two sequences of events.
- An electron is transferred from the metal to the substrate to give a radical anion.
A second electron transfer to the radical anion occurs to give a closed-shell dianion.
The dianion is protonated to give a close-shell anion.
or
- An electron is transferred from the metal to the substrate to give a radical anion.
The radical anion is pronated to give a neutral radical.
A second electron transfer occurs to give a closed-shell anion.
The solvent is usually liquid ammonia, in which the electrons from the metal are solvatedd, giving a dark blue solution. Dissolving metals have been used for the conjugate reduction of α,β-unsaturated ketones for a very long time, and the method is still one of only a few good ones for accomplishing this transformation.
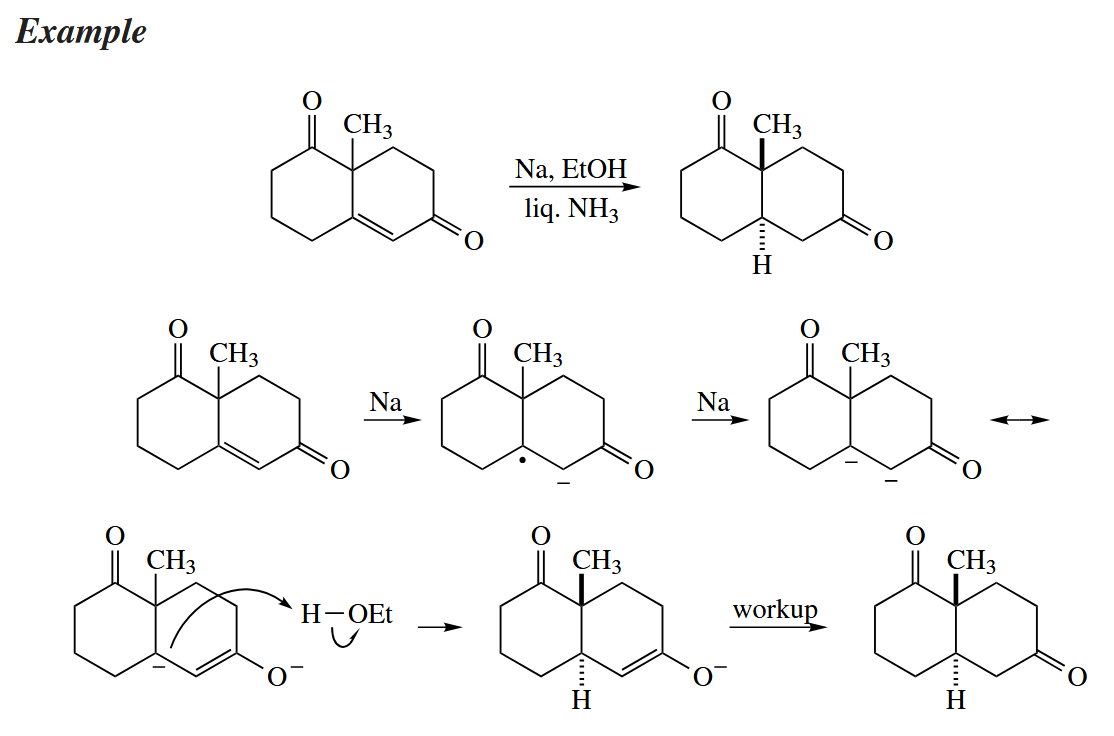
Saturated ketones are reduced to the thermodynamically more stable alcohols in the Bouveault-Blanc reduction by an almost identical mechanism.
In the reduction of alkynes to trans alkenes, the second electron transfer occurs after the first protonation.
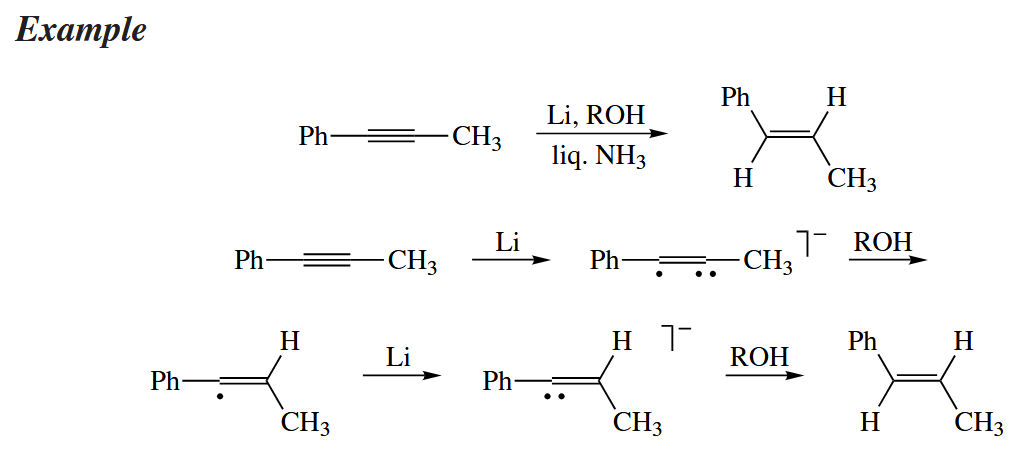
The Birch reduction converts arenes to 1,4-cyclohexadienes. The protonation steps determine the regiochemistry of the reduction.
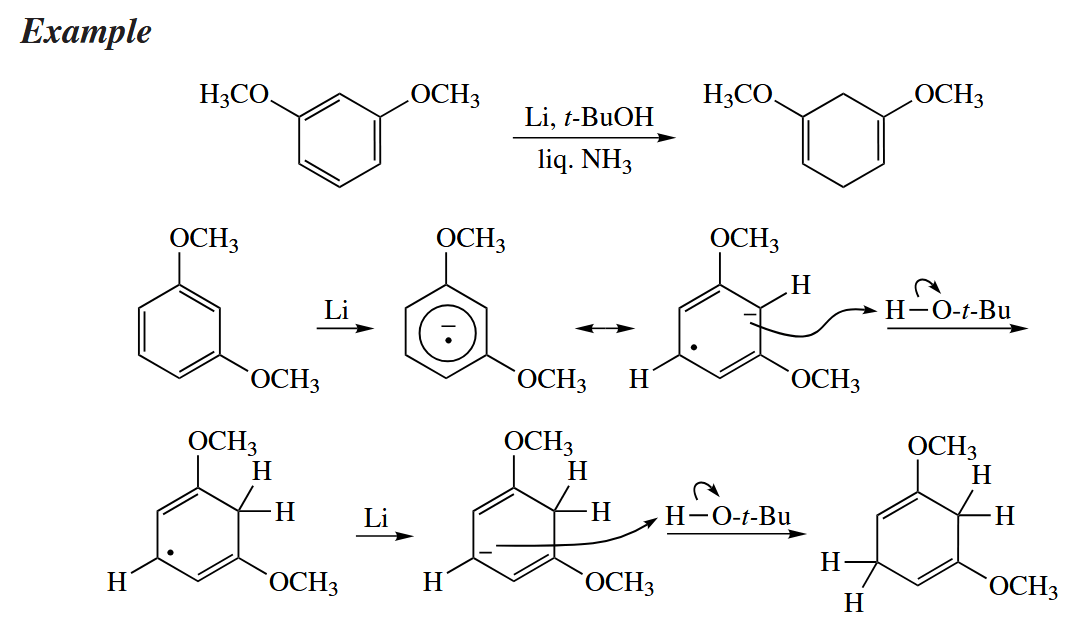
Birch reductions are highly intolerant of functionality. All benzylic ethers, alcohols, and carbonyl groups are reduced away and replaced with H, as are halides anywhere in the compound. The reductive cleavage of benzylic ethers makes the benzyl group a useful protecting group for alcohols.
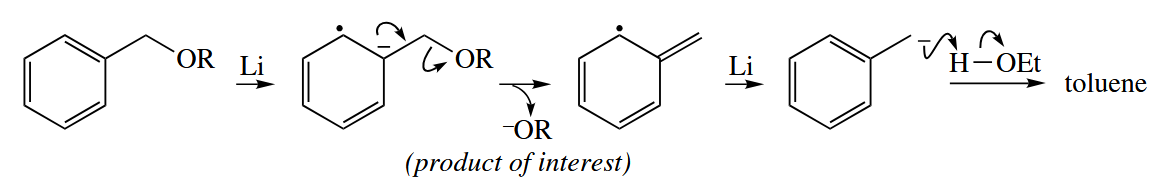
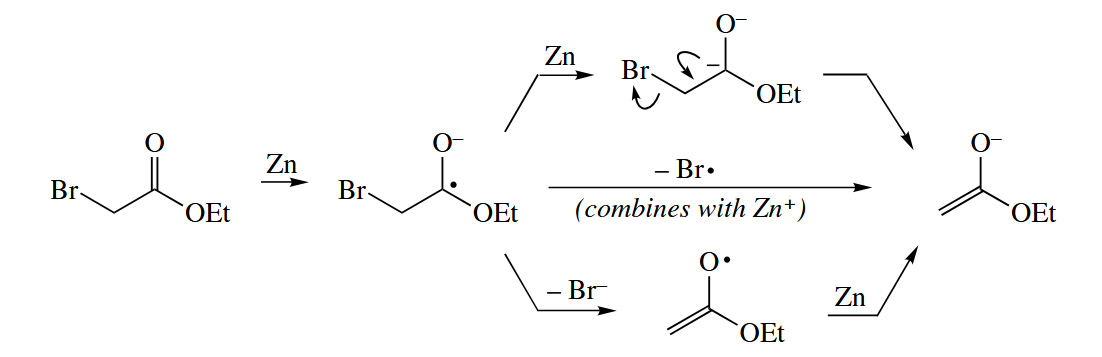
Leaving groups on the α-carbon of carbonyl compounds (e.g., Br and OR) are reduced away by one-electron reducing agents. α-Bromocarbonyl compouds are reduced to the corresponding enolates by Zn in the Reformatsky reaction. After the initial electron transfer, several pathways are possible, but all lead to the enolate.
Samarium diiodide (), an oxophilic, one-electron reducing agent, is particularly useful for the α-deoxygenation of ketones. It transfers one electron to a C=O π bond to give the ketyl.
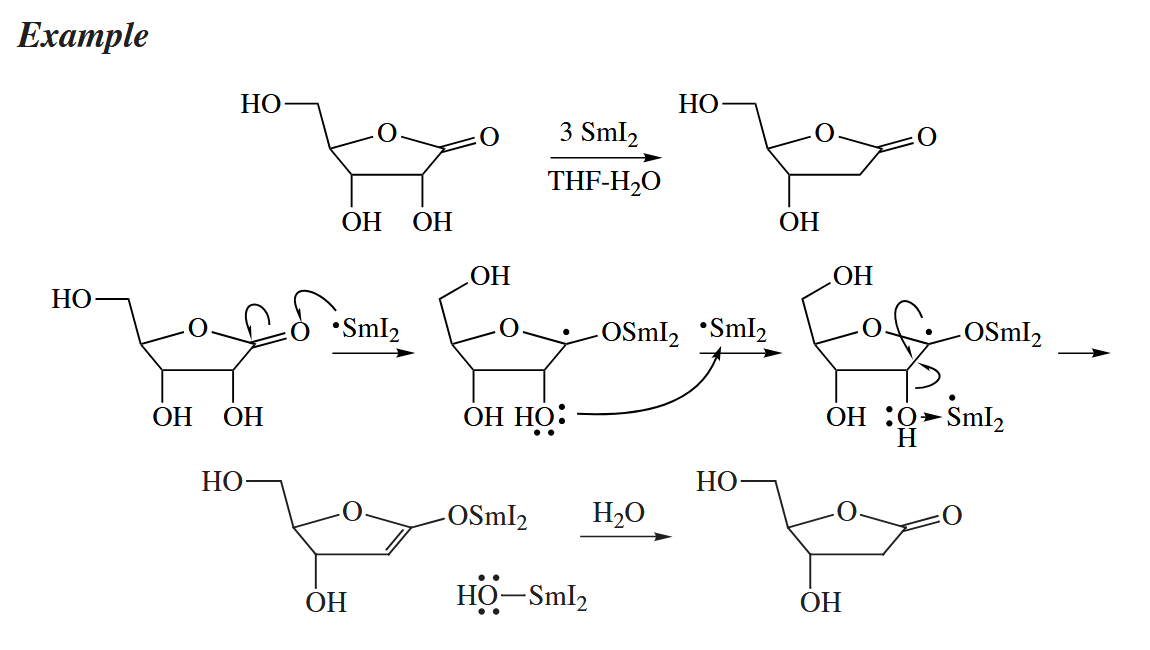
In this example, three equivalents of are used in order to provide an excess of the reagent.
In the pinacol coupling, two ketones are reductively coupled to give a 1,2-diol. The acyloin condensation converts two esters to an α-hydroxyketone, often in an intramolecular fashion.

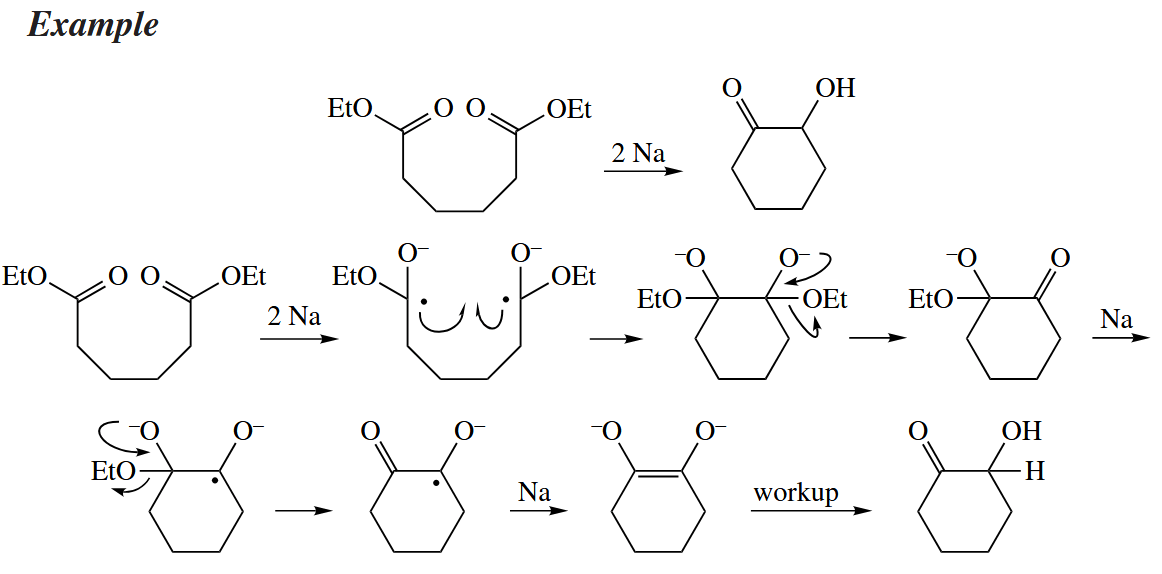
Modern acyloin condensation are usually executed in the presence of , and a bis(silyoxy)alkene is obtained as the immediate product.
The p-methoxyphenyl group can be removed from N or O by a one-electron oxidation promoted by CAN or DDQ. The aryl group is further oxidized to the quinone after it is cleaved from the substrate, so at least two equivalents of oxidant is required.
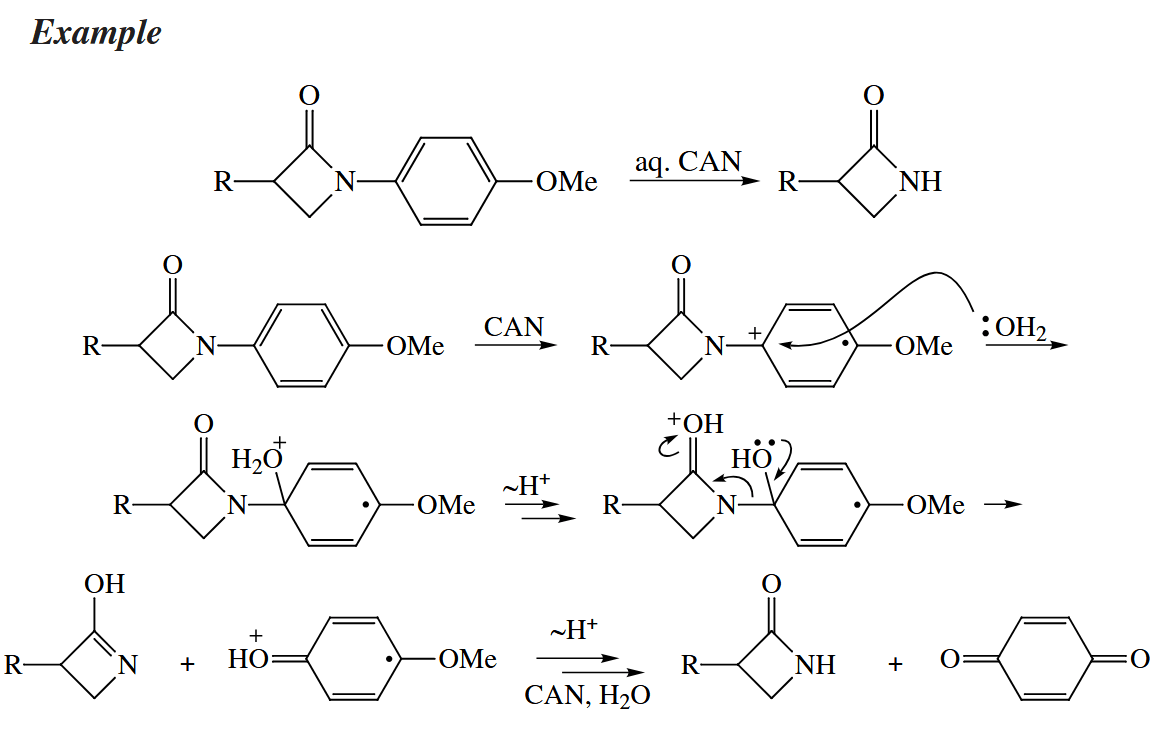
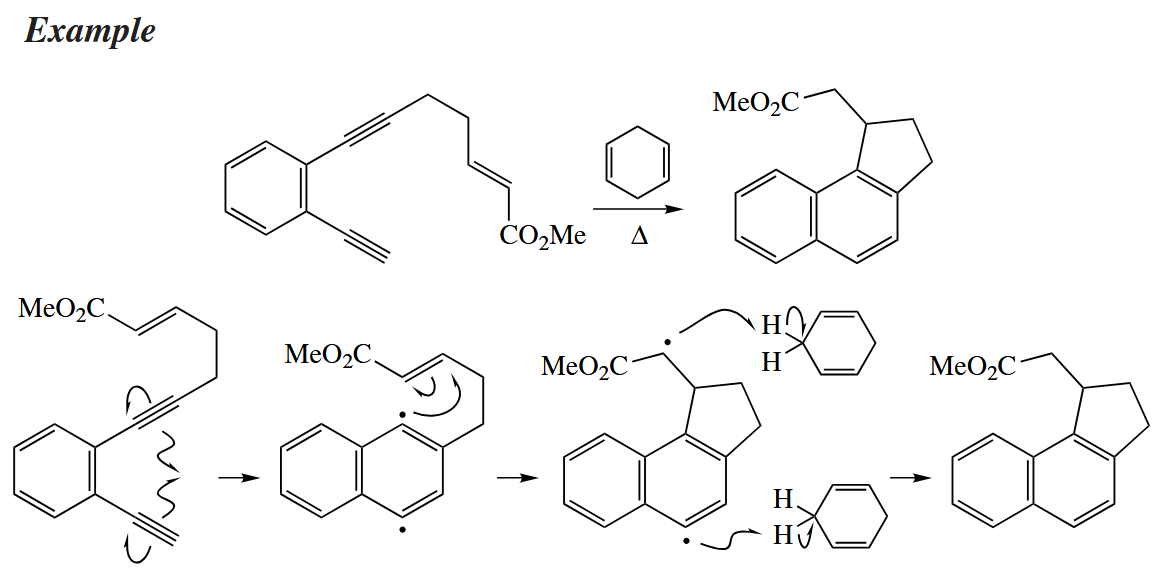
5.3.3 Cycloaromatizations
The synthetic potential of cycloaromatizations has only just begun to be explored. The two aryl radicals that are generated in the cycloaromatization are usually trapped by abstraction of H· from a compound such as 1,4-cyclohexadiene.
5.4 Miscellaneous Radical Reactions
5.4.1 1,2-Anionic Rearrangements; Lone-Pair Inversion
The Stevens rearrangement and the Wittig rearrangement (nonallylic version) can be classified as four-electron [1,2] sigmatropic rearrangements.
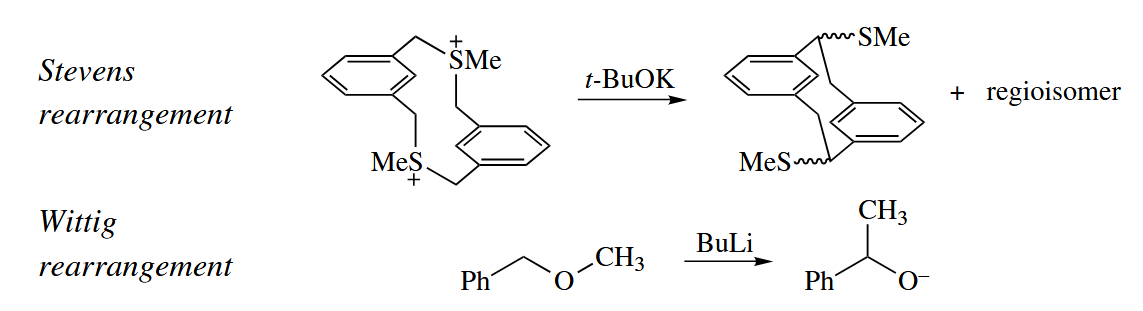
These two reactions proceed can be explained by a two-step, nonconcerted mechanism involving free radical. So the Woodward-Hoffmann rules is not applicable to the reactions.
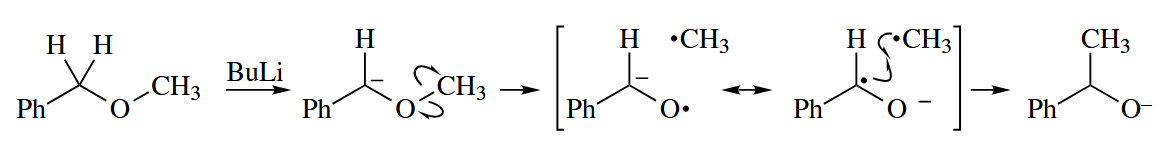
5.4.2 Triplet Carbenes and Nitrenes
Upon photolysis of a diazo compound or an azide, a triplet carbene or nitrene is generated. The two unshared electrons in triplet carbenes have aligned spins and reside in different orbitals.
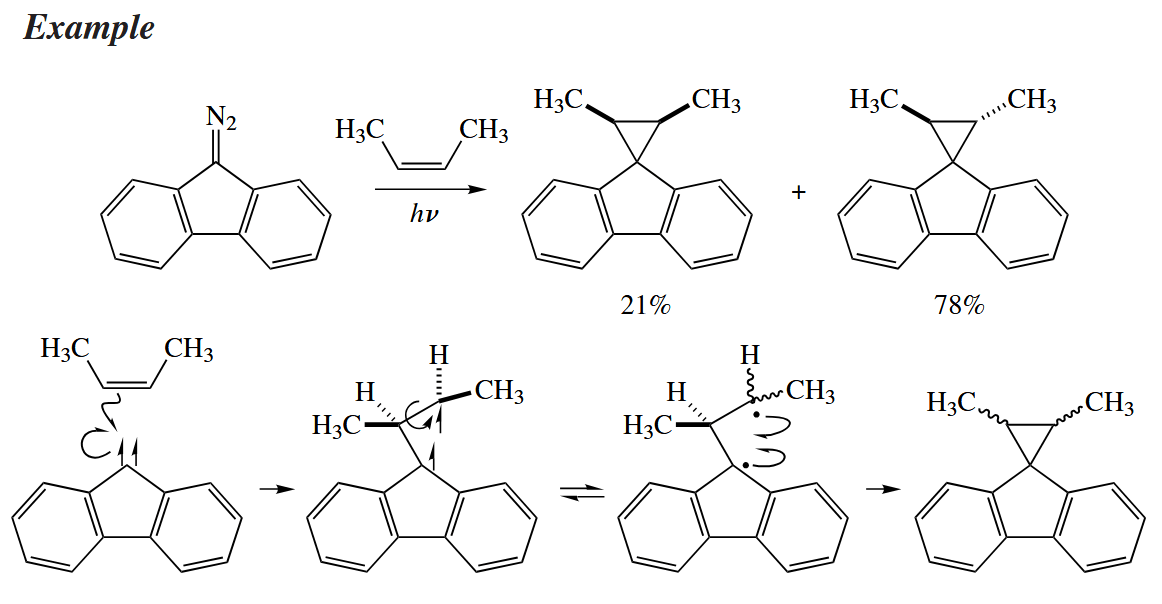
However, singlet carbenes are more useful synthetically because they react stereospecifically. Most carbene-generating techniques give singlet carbenes.
5.5 Summary
Free radicals are seven-electron, electron-deficient species. Many free radicals proceed by chain mechanisms, but photochemical rearrangements, metal-mediated oxidation and reductions, and cycloaromatizations do not. 自由基是七个电子的缺电子物种,许多自由基反应通过链式机理进行,但是光化学重排,金属介导的氧化/还原和环化芳构化例外。
Free radicals undergo eight typical reactions, but three are most important: atom abstraction, addition to a π bond, and fragmentation. 自由基共有八种经典的反应,其中三种最为重要的反应是:原子捕获,对π键的加成和裂解反应。
Carbocations are six-electron, electron-deficient species. They undergo three typical reactions: addition of a nucleophile, fragmentation, and rearrangement by a 1,2-shift. 碳正离子是六个电子的缺电子物种,它的三个经典反应是:亲核试剂加成,裂解反应和1,2-重排。
Carbenes are also six-electron, electron-deficient species. They undergo four typical reactions: cyclopropanation, insertion into a C-H bond, addition of a nucleophile, and rearrangement by a 1,2-shift. 卡宾也是六个电子的缺电子物种,它的四个经典反应是:环丙基化,C-H键插入,亲核试剂加成和1,2-重排。
The similarities that exist among free radicals, carbocations, and carbenes are apparent. The differences among them are due to their different electron counts and the presence or absence of an unshared pair of electrons. 自由基、碳正离子和卡宾之间有很多相似之处,而它们之间的不同主要是电子数不同和是否有成对电子导致的差异。
Chapter V
2. The production of chlorofluorocarbons, or CFCs, has been banned by international treaty because of their deleterious effect on the ozone layer. The ozone layer absorbs much of the Sun’s dangerous UV radiation before it reaches the Earth’s surface. CFCs are extremely stable in the lower atmosphere (one reason why they are so useful), but when they reach the stratosphere they decompose, producing potent catalysts of ozone destruction. Ozone destruction is most evident above Antarctica during the spring, when this region is exposed to the Sun for the first time in months. Dichlorodifluoromethane () is a typical CFC. Hydrochlorofluorocarbons, or HCFCs, are being promoted as temporary replacements for CFCs. HCFCs, unlike CFCs, have at least one C–H bond. HCFCs are not less prone than CFCs to decompose in the stratosphere, but there is a pathway by which HCFCs can decompose in the lower atmosphere (where they cannot damage ozone) that is not accessible to CFCs. 2,2,2-Trichloro-1,1-difluoroethane, , is a typical HCFC.

Ans

CFC,即氟氯烃,一个更广为人知的名字是氟利昂。早些年间被广泛用于空调制冷剂,但是随着人们意识到其对臭氧层的危害,最终被禁止使用。CFC对臭氧层的危害主要体现在CFC受到紫外光照射激发时会断裂产生Cl自由基,氯自由基会作为催化剂不断催化臭氧分解,因此对臭氧层有相当显著的危害。
一种替代方案是使用HCFC,相比于一般的CFC,HCFC在分子结构上多出一个C-H键,这使得它在更低的高度就会被夺氢产生碳自由基,并进行其他转化,不会危害到臭氧层。
3. Draw mechanisms for each of the following reactions.
_c42119651ed2b.png)
Ans
_b98aad829ae87.png)
本题为烷基取代的 Birch 还原,3当量的锂中,一份用于中和羧酸生成羧酸锂盐,剩余两份用来完成 Birch 还原。注意取代基对 Birch 还原区域选择性的影响。反应中生成的自由基阴离子会对溴化物进行亲核取代,然后再完成剩余的还原。
_ba7b9e3789273.png)
Ans
_4be1f6aab8114.png)
根据题目中提示,两种反应路径都会得到同一种四元环产物,再加上副产物的结构,容易发现第二种反应路径的副产物是第一种路径的副产物进一步裂解得到的。
首先根据光照条件,可得出应该是酮羰基被激发,后经历 Norrish type II 反应碎裂成两个部分,即为第一种反应路径。第二种路径可根据苯甲醛产物推测出碳自由基捕获了H原子,因而可推得反应机理。
_633abce73fa46.png)
Ans
_97c8acdb501b6.png)
观察产物发现在原料中羰基碳和四级碳之间的键断裂了,因此可推出发生了 Norrish type I 反应。第一步是光照条件下羰基被激发,随后发生 Norrish type I 反应,然后根据产物结构可推出剩下的反应机理。
_53487a65302ee.png)
Ans
_c9202a731f64f.png)
本题为自动氧化过程,注意氧气既是引发剂,又参与了氧化反应。由于氧气是双自由基,因此在发生自由基结合之后仍然可以捕获 PhSH 的质子完成链转移。
_ddb86fb94eaf1.png)
Ans
_420ce2b6eb80d.png)
本题类似于 Barton-McCombie 反应,只不过本题中不是黄原酸酯而是硝基。由于位阻因素,所以三丁基锡自由基难以直接进攻 N 原子,其先进攻 O 原子再发生断裂实现硝基的脱除。
_b0bf639d084f3.png)
Ans
_a28e9f105c120.png)
本题的原料硫代异羟肟酸酯(也称 Barton ester)实际上是 Barton 脱羧反应的中间产物,最弱的 C=S 双键首先被光照激发,随后弱的 N-O 键断裂给出羧酸自由基,随后发生脱羧和分子内环化,最后捕获溶剂的氯原子。反应也可以看作链式机理。
_e06b8c03025df.png)
Ans
_664fd2c84c693.png)
产物一是底物脱去醛基后得到,易于写出机理。产物二经历了一个三元环中间体的加成-断裂过程,表观上体现为交换了碳的位置。同时需要注意完成反应的链转移过程。
_a5146b9170e25.png)
Ans
_17acff8d15acb.png)
加热过程会断裂反应物中最弱的 O-O 键,然后临近的键断裂后再结合得到二酰基过氧化物中间体。该中间体如果脱去一分子二氧化碳则得产物二,脱去两分子二氧化碳则为产物一。
_b034c5f29e950.png)
Ans
_5e938582a9afa.png)
本题为 Barton 反应,关键步骤为光照断裂 N-O 键脱去 NO 分子,然后氧自由基通过六元环过渡态攫氢,新生成的自由基再与 NO 分子结合。
_125f0bb39bdf0.png)
Ans
_0f212260ff793.png)
本题的第一个难点在于正确标号,确定出断裂的那根键。以图示为例,确定了断裂键为 C3-C7 后,就可以开始写机理了。
反应试剂和 Birch 还原试剂相同,因此是单电子还原,首先还原羰基得到自由基阴离子,断裂临近的碳碳键后再转移一次电子,并实现质子化,最后实现互变异构。
_bfeb9fcba6bc4.png)
Ans
_b82c1b575caa2.png)
本题和(aa)的底物相同,但是使用不同的试剂就得到了不同的产物,首先仍是标号,不过本题的标号相对上一题更为清晰,表观上可以认为处在下方的 C4 插入了平面的六元环。
本题为链式机理,链转移的第一步非常显然,后续可以认为有不同的反应路径,都经历了一些高张力的结构,根据第一步标号分析的成键、断键一步步写即可。
_aa4e391149b25.png)
Ans
_2935e639b3177.png)
题干提示硝酸铁和 CAN (即硝酸铈铵)具有相同反应性,因此可知其为单电子氧化剂。它会夺去反应物中能量最高的电子,即硅醚氧的一个孤对电子,随后临近三元环断裂生成烷基自由基,并发生分子内环化。反应体系中的1,4-环己二烯为质子源。
_c32e819aab9cc.png)
Ans
_3b1e5ed7235b2.png)
本题中醋酸锰首先与羰基结合,将其转变为烯醇盐,随后氧化,发生分子内的串联自由基环化,一步将四个六元环关闭。最后醋酸铜也作为单电子氧化剂给出碳正离子并消除得到产物。
本章的自由基反应模式相对固定,常见的几种引发剂、链引发、链转移过程、几个典型的反应掌握后大多数反应都能顺利完成,自由基中间体和碳正离子中间体有一定的相似之处,因此可以借助极性反应的机理辅助理解。但是仍有不同之处,例如自由基反应中羰基旁边的碳碳键易于断裂,而极性机理中极少见。
第五章结束。